Page 2 of 16
FM13.3 | Poisons: Classification, Toxicokinetics & Diagnosis — SDL Guide
Learning Objectives
- Classify poisons by origin (animal, vegetable, mineral, synthetic) and by action (corrosive, irritant, neurotic, cardiac, asphyxiant) per the Indian forensic medicine classification scheme
- Describe toxicokinetics — ADME (absorption, distribution, metabolism, excretion) — as applied to poisoning scenarios, including volume of distribution and first-pass effect
- Explain toxicodynamics: dose-response relationships, receptor interactions, tolerance, idiosyncrasy, and synergism
- Describe the clinical approach to diagnosing poisoning in the living and the post-mortem approach in the dead
INSTRUCTIONS
Before you can manage any poisoning or write a credible medico-legal report, you must be able to classify the offending agent and understand what it does in the body. Classification is not a memory exercise — it is the reasoning framework that takes you from a patient's clinical picture to the likely poison class, directs your immediate management, and determines which IPC section and FSL analysis protocol apply. Toxicokinetics explains why the same dose kills one patient and leaves another intact. This module gives you that dual framework.
References
- KSN Reddy — Essentials of Forensic Medicine & Toxicology, 34th ed. (textbook)
- BV Subrahmanyam — Modi's Medical Jurisprudence and Toxicology, 24th ed. (textbook)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
Two patients arrive in the emergency department within minutes of each other. The first is a 45-year-old housewife with burns around her mouth, severe throat pain, and drooling of frothy secretions — her daughter says she 'swallowed toilet cleaner.' The second is a 19-year-old student who is drowsy, has constricted pupils, and slow shallow breathing — his friend says he was at a party. Both are poisoned, but the immediate management, the FSL samples required, and the IPC sections applicable differ completely. Without a systematic classification framework and understanding of how each poison behaves in the body, the treating physician is guessing.
WHY THIS MATTERS
Poison classification is used every day in Indian emergency medicine and forensic practice. The clinical toxidrome — the constellation of signs produced by a poison class — allows a trained physician to identify the likely agent before the laboratory returns results, enabling immediate targeted management. In medico-legal contexts, the classification of a poison determines which section of the Poisons Act or NDPS Act applies, which analytical method the FSL will use, and how the medico-legal report will categorise the mode of poisoning. Understanding toxicokinetics explains variability in outcome — why an agricultural worker with chronic OP exposure tolerates a dose that would kill a naive individual.
RECALL
From Year-1 Pharmacology: you already know ADME — absorption, distribution, metabolism, and excretion — as the four processes governing drug behaviour. You recall that hepatic first-pass metabolism reduces bioavailability of orally administered drugs, and that CYP450 enzymes are central to Phase I metabolism. From Biochemistry, you understand enzyme kinetics and the concept of saturable pathways. Today we apply this framework to poisons, where the same processes determine how quickly a lethal concentration builds in tissues and how the body attempts (often unsuccessfully) to eliminate the toxin.
Classification of Poisons: By Origin and By Action
The classification of poisons provides an ordered system for approaching the vast chemical universe of toxic substances encountered in Indian forensic and clinical practice. Two complementary axes — classification by origin and classification by action — together define the two-dimensional framework used in Reddy's Essentials and Modi's Medical Jurisprudence, which are the authoritative Indian references. Both axes must be understood because they serve different purposes: origin-based classification directs the FSL analyst to the appropriate analytical method, while action-based classification guides the clinician to the correct antidote and management strategy.
Classification by origin divides poisons into four groups. Animal poisons (venoms and toxins of biological animal origin) include snake venoms, scorpion venom, bee/wasp venom, and marine toxins. Their composition is complex — proteins, enzymes, and peptides — making them analytically challenging. Vegetable poisons (plant-derived alkaloids, glycosides, and resins) include some of India's most important forensic poisons: aconitine (Aconitum species — often used in homicide), strychnine (nux vomica), abrin (Abrus precatorius — rosary pea, commonly available), ricin (Ricinus communis — castor), and digitaloid glycosides (oleander). Mineral poisons include metals and their salts (arsenic, lead, mercury, copper, iron, barium) as well as inorganic acids and alkalis. Arsenic remains the most important mineral poison in Indian forensic practice owing to its historical use and widespread environmental presence. Synthetic poisons encompass organophosphate and carbamate pesticides (leading cause of poisoning mortality in rural India), organochlorine pesticides, solvents (methanol, carbon tetrachloride), and pharmaceutical drugs in overdose.
Classification by action is the more clinically relevant axis. Corrosive poisons — strong mineral acids (sulphuric acid, hydrochloric acid, nitric acid) and strong alkalis (sodium hydroxide, potassium hydroxide, carbolic acid/phenol) — destroy tissue on contact by chemical necrosis. They produce burns at the lips, oral mucosa, oesophagus, and stomach. Their medico-legal significance is high because the injuries are severe, visible, and immediately attributable. Irritant poisons cause inflammation and irritation, either locally (at the site of contact — skin, GI mucosa) or remotely (systemic organs). They are subdivided into local irritants (strong acids in lower concentration, plant irritants) and remote systemic irritants (arsenic, antimony, mercury — causing GI inflammation and systemic organ damage). Neurotic poisons act primarily on the nervous system and are further subclassified: spinal irritants (strychnine — causes tetanic convulsions), cerebral depressants (ethanol, chloroform, opioids — sedation and coma), cerebral excitants (cocaine, amphetamines), and peripheral neurotoxins (curare-type neuromuscular blockers, cobra neurotoxins). Cardiac poisons act on myocardial conduction and contractility — digitalis glycosides, aconitine, and oleander are the key examples, producing life-threatening dysrhythmias. Asphyxiant poisons cause death by preventing oxygen delivery or utilisation: simple asphyxiants (CO₂, methane — dilute ambient oxygen without direct toxic effect) and chemical asphyxiants (carbon monoxide — binds haemoglobin with 240× the affinity of oxygen; cyanide — inhibits cytochrome c oxidase, blocking cellular respiration). In addition, poisons may be classified by target organ (hepatotoxic — paracetamol, phosphorus, CCl₄; nephrotoxic — ethylene glycol, mercuric chloride; haematotoxic — arsine, lead, naphthalene).
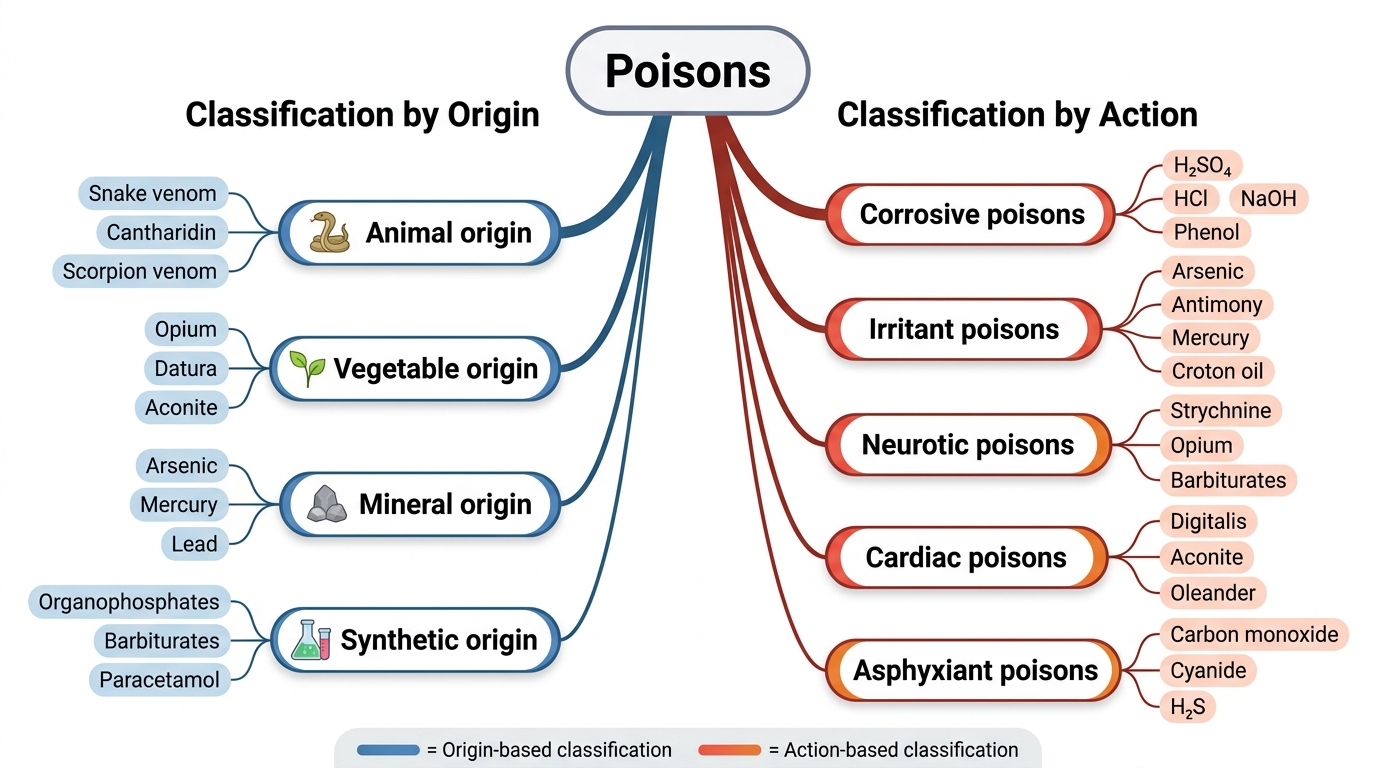
Two-Axis Classification of Poisons
| Classification by Action | Mechanism | Key Examples | Typical Presentation |
|---|---|---|---|
| Corrosive | Chemical necrosis on contact | H₂SO₄, HCl, NaOH, phenol | Perioral burns, haematemesis, stricture |
| Irritant (local) | Mucosal/skin inflammation | Croton oil, Calotropis | GI pain, vomiting, diarrhoea |
| Irritant (remote/systemic) | Systemic organ inflammation | Arsenic, antimony, mercury | GI + hepatorenal toxicity |
| Neurotic (spinal) | Strychnine-like convulsions | Strychnine, brucine | Tetanic spasms, opisthotonos |
| Neurotic (cerebral depressant) | CNS depression | Opioids, barbiturates, ethanol | Sedation, coma, respiratory depression |
| Cardiac | Dysrhythmia / conduction block | Digitalis, aconitine, oleander | Bradycardia, heart block, VF |
| Asphyxiant (chemical) | Blocks O₂ use/transport | CO, cyanide | Cherry-red cyanosis (CO), bitter almond odour (HCN) |
SELF-CHECK
A 60-year-old farmer is found dead in his field. Post-mortem shows cherry-red lividity, bright red blood, and no other external injuries. The FSL will most likely use which analytical method to confirm the suspected poison?
A. Thin layer chromatography (TLC) for alkaloid detection
B. Atomic absorption spectroscopy (AAS) for arsenic
C. Co-oximetry / spectrophotometric analysis of blood for carboxyhaemoglobin
D. Gas chromatography (GC) for volatile organic compounds
Reveal Answer
Answer: C. Co-oximetry / spectrophotometric analysis of blood for carboxyhaemoglobin
Cherry-red lividity with bright red blood is pathognomonic of carbon monoxide (CO) poisoning — carboxyhaemoglobin produces the distinctive colour. The confirmatory test for CO is spectrophotometric analysis (co-oximetry) of blood to measure carboxyhaemoglobin saturation. TLC detects alkaloids; AAS detects metallic poisons; GC detects volatile organic compounds — none of these is the primary method for CO confirmation.
Toxicokinetics: ADME in Poisoning
Toxicokinetics describes what the body does to a poison — how it is absorbed, distributed through tissues, chemically transformed (metabolised), and eliminated. The same ADME framework that governs therapeutic drugs applies to poisons, but the clinical stakes are inverted: instead of optimising efficacy, the goal is to understand how to slow absorption, accelerate elimination, and prevent toxic concentrations from accumulating at target organs.
Absorption is the process by which a poison enters the systemic circulation. The route of absorption critically determines the speed of onset and peak concentration. Oral ingestion is the most common route in poisoning — the GI tract's large surface area, combined with the rich blood supply of the small intestine, allows rapid absorption of most organic compounds. Inhalation is the fastest route to systemic effect (bypasses GI transit and partially bypasses hepatic first-pass), which is why CO, cyanide gas, and methanol vapour cause rapid clinical deterioration. Transdermal absorption is slow for most substances but highly relevant for lipid-soluble organophosphates (which penetrate agricultural workers' skin through prolonged field exposure) and for nicotine. Parenteral injection bypasses all absorption barriers and delivers the full dose to the bloodstream immediately.
For oral poisons, the first-pass effect is a critical modifying factor. Absorbed drug passes from the intestinal lumen via the portal circulation to the liver before reaching the systemic circulation. Hepatic enzymes (principally CYP450 family — CYP3A4, CYP2D6) may substantially metabolise the poison before it reaches target organs. A high first-pass effect reduces systemic bioavailability, which partly explains why the same oral dose is less toxic than an IV dose. However, first-pass effect can also generate toxic metabolites: paracetamol is metabolised to the reactive intermediate NAPQI (N-acetyl-p-benzoquinone imine), which causes hepatocellular necrosis in overdose.
Distribution describes how a poison is transported from the bloodstream to tissues. The volume of distribution (Vd) is a mathematical construct — expressed in L/kg — representing the apparent volume into which a drug distributes if it were uniformly distributed at the plasma concentration. A low Vd (~0.1 L/kg, e.g., warfarin) indicates the poison remains largely in the plasma compartment and is accessible to haemodialysis. A high Vd (>1 L/kg, e.g., chloroquine at 250–800 L/kg) indicates extensive tissue binding; the plasma concentration is a tiny fraction of the total body burden, and haemodialysis will not significantly reduce the total amount of poison in the body. Plasma protein binding also affects distribution: highly protein-bound poisons (>90%) have a small free fraction available for toxic action, but they are not readily dialysed and can be displaced by competing drugs, suddenly increasing the free (toxic) fraction.
Metabolism (biotransformation) modifies the chemical structure of a poison, usually rendering it more water-soluble for excretion. Phase I reactions (oxidation, reduction, hydrolysis — predominantly hepatic CYP450) create polar intermediates, sometimes more toxic than the parent compound (toxication). Phase II reactions (conjugation with glucuronide, sulphate, glycine, glutathione) generate water-soluble conjugates that are readily excreted. Saturation of Phase II pathways — as occurs in paracetamol overdose when glutathione is depleted — allows accumulation of toxic intermediates.
Excretion removes the poison (or its metabolites) from the body. The kidney is the primary excretory organ for water-soluble compounds. Renal excretion depends on: filtration (affected by protein binding), tubular secretion (active transport), and tubular reabsorption (affected by urinary pH — the basis of forced diuresis). Lipid-soluble compounds (e.g., volatile organics) are excreted by the lungs. Biliary excretion applies to large molecular weight compounds and may result in enterohepatic recirculation, prolonging the toxic effect.
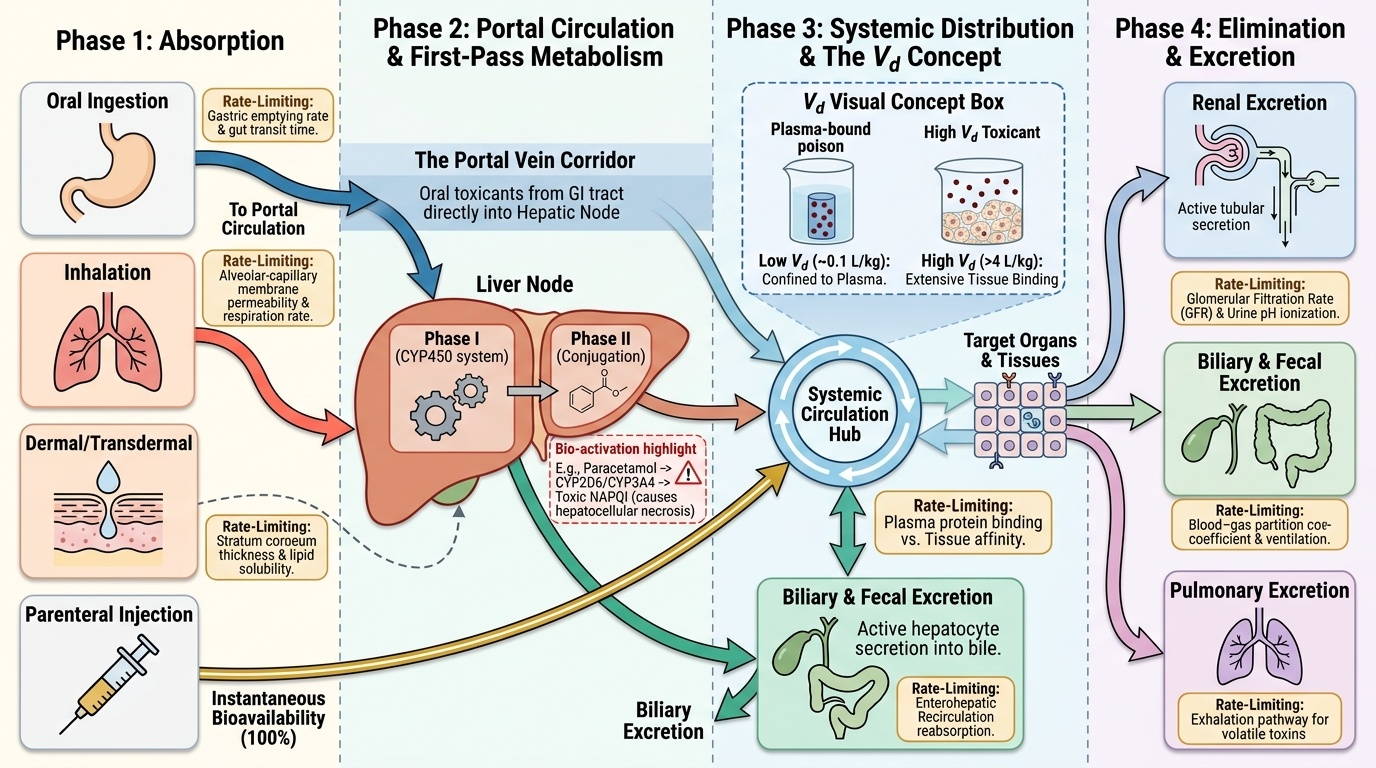
Provided image
Key toxicokinetic concepts:
• First-pass effect — reduces oral bioavailability; can generate toxic metabolites (e.g., paracetamol → NAPQI)
• Volume of distribution (Vd) — low Vd = confined to plasma (dialysable); high Vd = tissue-bound (dialysis ineffective)
• Protein binding — high binding reduces free fraction and dialysability; displacement can precipitate toxicity
• Phase I metabolism (CYP450) — may generate reactive toxic intermediates
• Phase II metabolism (conjugation) — water-solubilises for excretion; saturable in overdose
• Urinary pH manipulation — alkaline urine traps weak acids (salicylates, phenobarbital) as ionised form, reducing reabsorption
Toxicodynamics: How Poisons Cause Harm
Toxicodynamics describes what the poison does to the body — the cellular and molecular mechanisms by which toxic injury occurs. Understanding these mechanisms allows the clinician to predict which organs are at risk, anticipate the clinical course, and rationally select antidotes that counteract the specific mechanism of harm.
The foundational concept is the dose-response relationship. At the molecular level, virtually all poisons exert their effects through interaction with specific biological targets — receptors, enzymes, ion channels, transport proteins, or structural macromolecules (DNA, membrane lipids). The relationship between dose and response follows a sigmoid curve: below a threshold dose, no measurable effect occurs; above threshold, effect increases steeply with dose; at very high doses the effect plateaus (receptor saturation or target saturation). The steepness of this curve (the 'slope') determines how rapidly increasing dose converts a therapeutic effect to a lethal one — this is directly reflected in the therapeutic index.
Several important pharmacodynamic phenomena alter the expected dose-response. Tolerance is a reduced response to the same dose following repeated exposure, requiring progressively higher doses to achieve the same effect. It underlies opioid dependence and the counterintuitive observation that a chronic opioid user tolerates a dose that would be fatal in an opioid-naive individual. Tolerance occurs through receptor downregulation, receptor desensitisation, or induction of metabolic enzymes. Idiosyncrasy is an abnormal, qualitatively different response that is genetically determined and unpredictable from the normal dose-response curve — for example, glucose-6-phosphate dehydrogenase (G6PD) deficiency causing haemolytic anaemia after exposure to primaquine or naphthalene (mothballs). Synergism occurs when two agents produce a combined effect greater than the sum of their individual effects — highly relevant in poly-drug poisonings (e.g., alcohol + benzodiazepines producing profound respiratory depression). Antagonism — the basis of antidote therapy — involves one agent reducing the effect of another, either through receptor competition (naloxone competitively antagonises opioid receptors), physiological antagonism (atropine counters muscarinic hyperstimulation from OPs), or chemical antagonism (chelating agents bind metal ions).
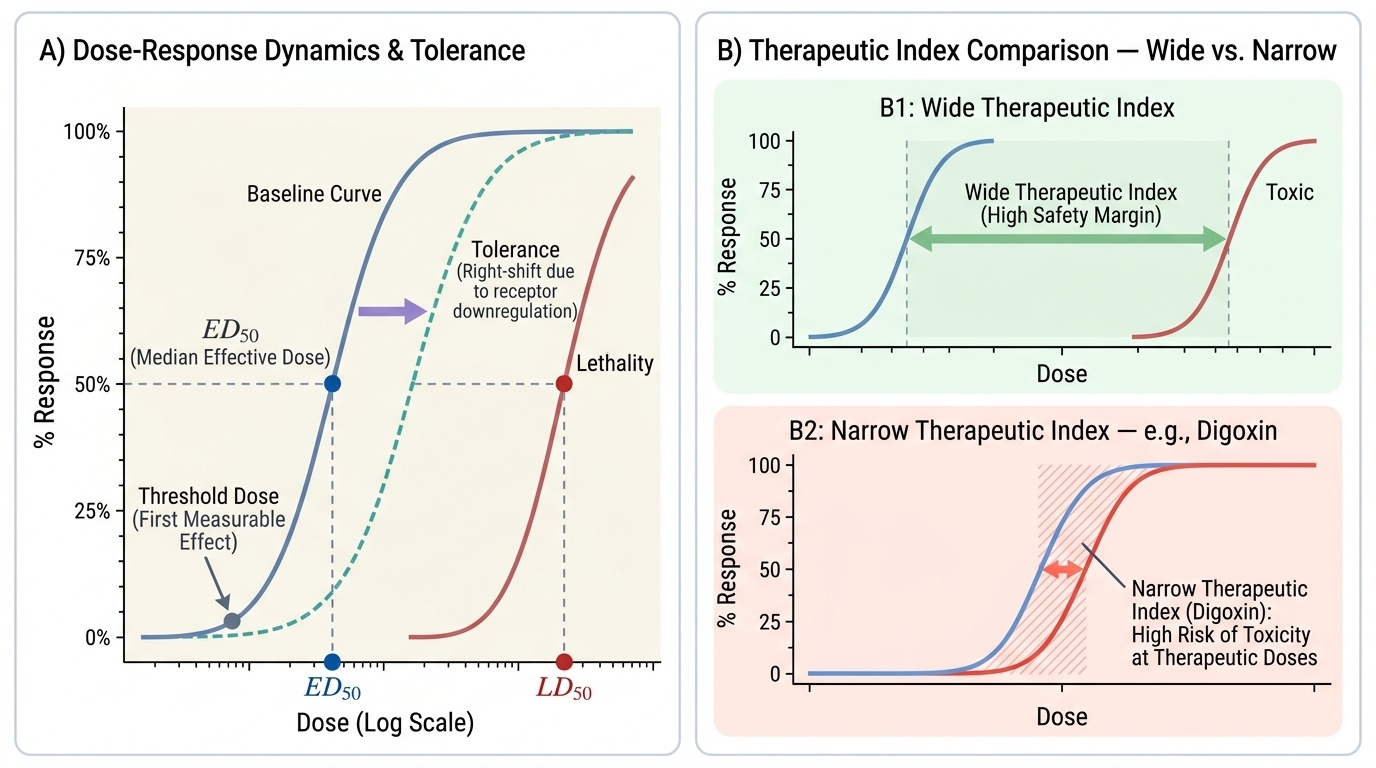
Provided image
Key mechanisms of lethal toxicity at the cellular level include:
• Ion channel disruption — tetrodotoxin blocks voltage-gated Na⁺ channels (nerve/muscle paralysis); aconitine activates Na⁺ channels causing cardiac dysrhythmia
• Mitochondrial toxicity — cyanide inhibits cytochrome c oxidase (Complex IV), blocking oxidative phosphorylation; metformin (in overdose) inhibits Complex I
• Enzyme inhibition — organophosphates irreversibly inhibit acetylcholinesterase, causing cholinergic excess; methanol is metabolised to formaldehyde → formate (inhibits cytochrome oxidase → metabolic acidosis)
• Membrane disruption — detergents and lipid-soluble solvents disrupt phospholipid bilayers
• Protein denaturation — strong acids and alkalis denature structural and enzymatic proteins on contact
• Reactive metabolites — paracetamol → NAPQI attacks hepatocellular proteins and lipids when glutathione stores are depleted