Page 2 of 25
IM1.1-7 | Heart Failure Foundations and Aetiology — SDL Guide (Part 2)
Compensatory Mechanisms in Heart Failure: Neurohormonal Adaptations
When cardiac output falls, the body mounts a series of short-term compensatory responses designed to restore perfusion. In the acute setting, these responses are adaptive and life-saving. However, when heart failure becomes chronic, the same responses become maladaptive — driving progressive ventricular remodelling, worsening the haemodynamic burden, and accelerating the disease process. Understanding these mechanisms is essential because they are the therapeutic targets of the drugs that save lives in heart failure: ACE inhibitors, angiotensin receptor blockers, beta-blockers, and mineralocorticoid receptor antagonists.
Provided image
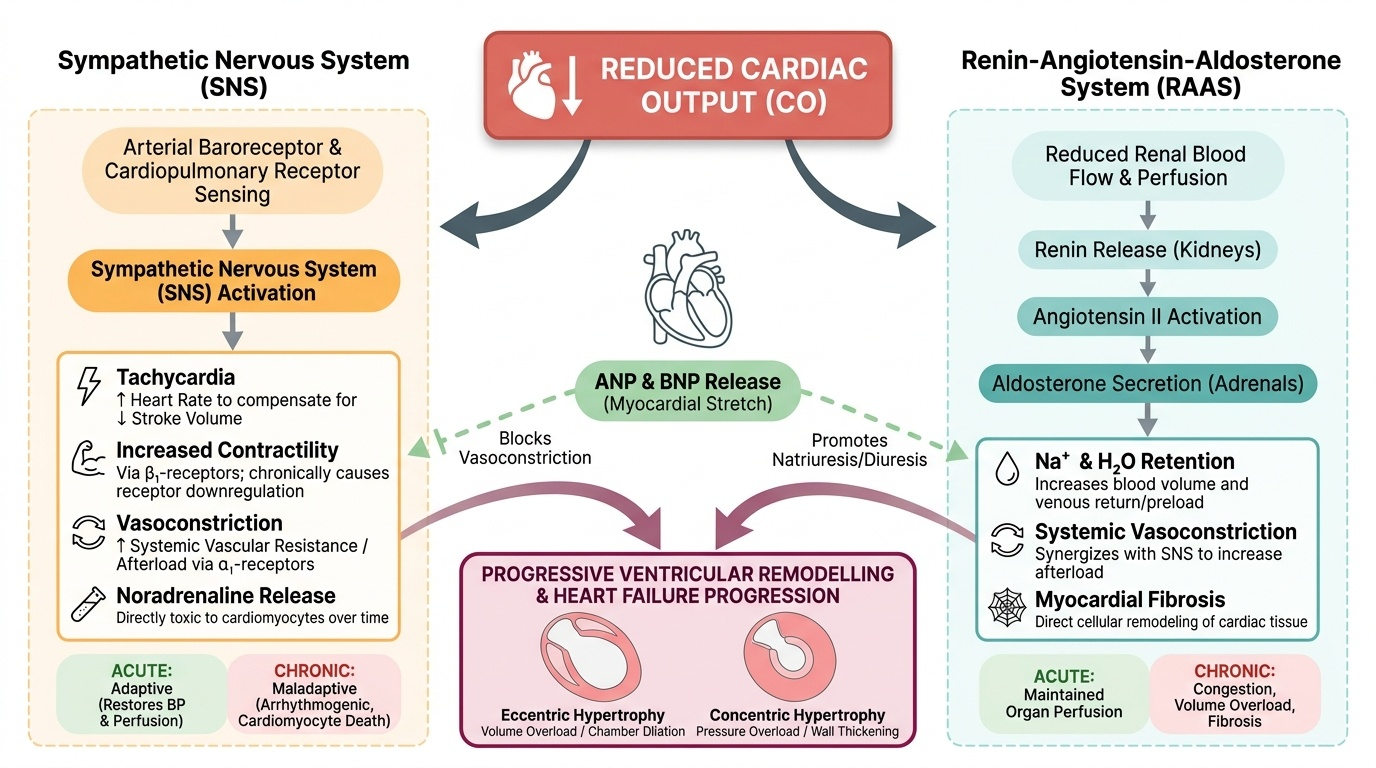
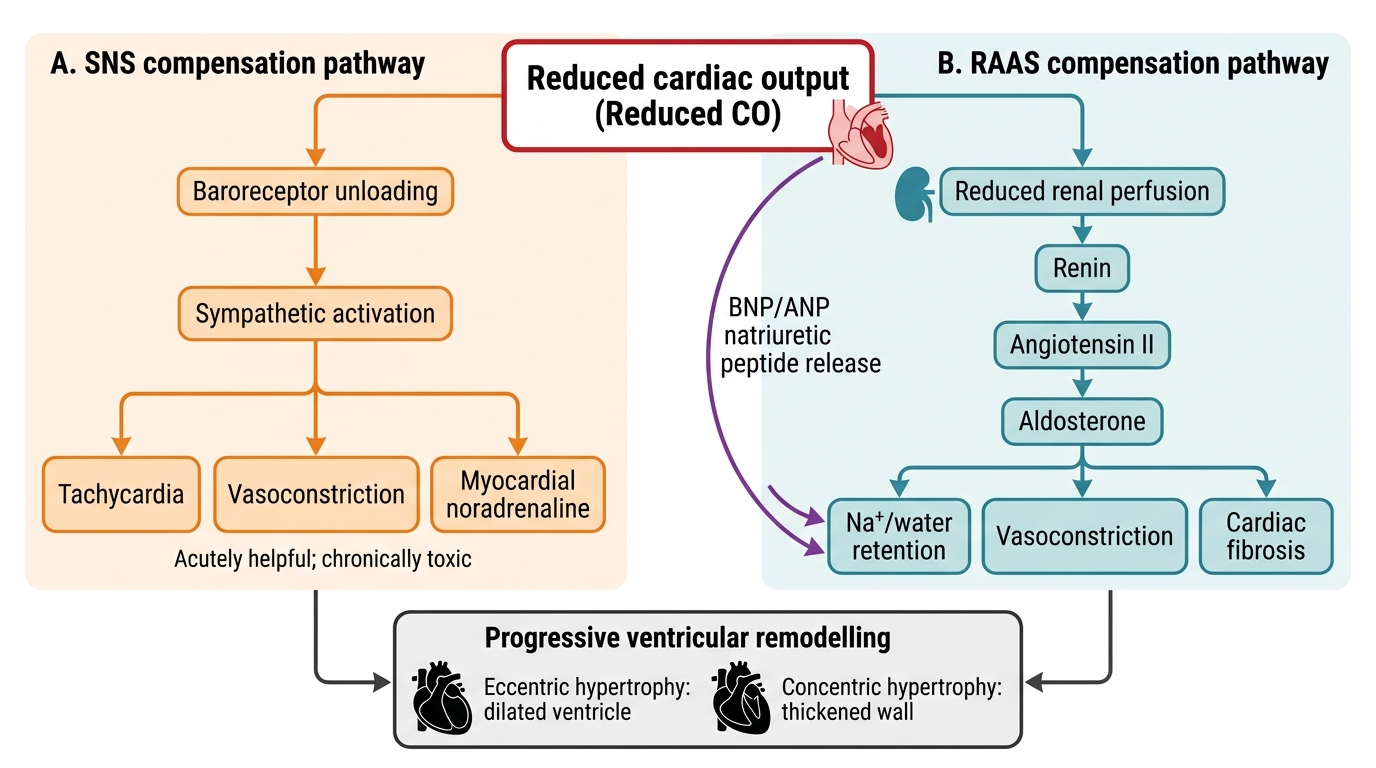
The three principal compensatory mechanisms are (1) the Frank-Starling mechanism, (2) neurohormonal activation (the renin-angiotensin-aldosterone system (RAAS) and the sympathetic nervous system (SNS)), and (3) cardiac remodelling.
1. Frank-Starling mechanism: A failing ventricle dilates in response to reduced stroke volume — the increased end-diastolic volume stretches sarcomeres and, within limits, increases contractile force (the ascending limb of the Frank-Starling curve). This mechanism transiently maintains cardiac output but at the cost of elevated filling pressures (causing the upstream congestion symptoms described above). On the descending limb of the Starling curve, further dilation actually reduces contractile efficiency — a point reached in severely dilated failing hearts.
2. Sympathetic nervous system activation: Reduced cardiac output is sensed by arterial baroreceptors and cardiopulmonary receptors in the failing chambers. The resultant withdrawal of parasympathetic tone and activation of the SNS causes:
- Tachycardia (increased HR to compensate for reduced SV) — acutely helpful, chronically energy-consuming and arrhythmogenic.
- Increased contractility via beta-1 adrenoceptor stimulation of the myocardium — acutely helpful; chronically causes beta-1 receptor downregulation and desensitisation, so the myocardium becomes less responsive to catecholamines.
- Vasoconstriction (increased afterload via alpha-1 receptor stimulation) — maintains blood pressure acutely; chronically increases the workload against which the failing ventricle must contract, further impairing stroke volume.
- Elevated circulating noradrenaline is directly toxic to cardiomyocytes, promoting apoptosis — chronic sympathetic activation is a key driver of progressive myocardial damage.
3. Renin-Angiotensin-Aldosterone System (RAAS) activation: Reduced renal perfusion (from low cardiac output and from SNS-driven renal vasoconstriction) activates the juxtaglomerular apparatus to release renin, which converts angiotensinogen to angiotensin I. Angiotensin I is converted to angiotensin II (Ang II) by angiotensin-converting enzyme (ACE) in the pulmonary vasculature. Ang II has multiple maladaptive effects in chronic HF:
- Systemic vasoconstriction → increased afterload.
- Aldosterone release from the adrenal cortex → sodium and water retention → volume overload → increased preload → pulmonary and systemic congestion.
- Direct cardiac and vascular fibrosis and hypertrophy — Ang II stimulates transforming growth factor-beta (TGF-β) and promotes collagen deposition → ventricular and vascular remodelling.
- Thirst stimulation via central Ang II receptors → fluid retention.
4. Natriuretic peptides — the counter-regulatory system: In response to elevated wall stress (raised filling pressures), the ventricles secrete brain natriuretic peptide (BNP) and its inactive precursor fragment NT-proBNP. These peptides counteract RAAS by promoting natriuresis (sodium excretion), vasodilation, and inhibiting aldosterone. BNP and NT-proBNP are the most important biomarkers in heart failure: elevated levels confirm the diagnosis, and their magnitude correlates with severity (NT-proBNP >125 pg/mL is the diagnostic cut-off; levels >450, >900, >1800 pg/mL for age groups <50, 50–75, >75 years correlate with severity). Despite natriuretic peptide release, the RAAS overrides this counter-regulation in established heart failure.
5. Cardiac remodelling: The chronic neurohormonal milieu drives structural changes in the heart — the process of ventricular remodelling. In HFrEF, the LV undergoes eccentric hypertrophy: chamber dilation with sarcomere replication in series, shifting the LV from its normal ellipsoid shape toward a spherical shape. This spherical geometry reduces ejection efficiency further (by the Law of Laplace: wall stress = pressure × radius / 2 × wall thickness — a dilated sphere generates greater wall stress for the same intracavitary pressure). In HFpEF, concentric hypertrophy predominates: pressure overload (from hypertension) drives sarcomere replication in parallel, thickening the wall without dilation but impairing relaxation and increasing stiffness. Both remodelling patterns reduce diastolic compliance and impair the heart's ability to adapt to changes in demand.
Neurohormonal Compensation in Heart Failure
SELF-CHECK
A 62-year-old man with ischaemic cardiomyopathy has an echocardiographic LVEF of 35%. He can walk on level ground for 200 metres but becomes breathless climbing one flight of stairs; he is comfortable at rest. What is his LVEF phenotype and NYHA functional class?
A. HFpEF, NYHA Class I
B. HFmrEF, NYHA Class II
C. HFrEF, NYHA Class II
D. HFrEF, NYHA Class III
Reveal Answer
Answer: C. HFrEF, NYHA Class II
LVEF 35% falls in the HFrEF range (≤40%). His symptoms appear on ordinary physical activity (climbing stairs) but he is comfortable at rest and can walk 200 m on level ground — this is slight-to-moderate limitation, consistent with NYHA Class II (ordinary activity causes dyspnoea). NYHA Class III would be marked limitation where less-than-ordinary activity causes symptoms. HFmrEF requires LVEF 41–49%. HFpEF requires LVEF ≥50%.
Factors That Exacerbate Heart Failure
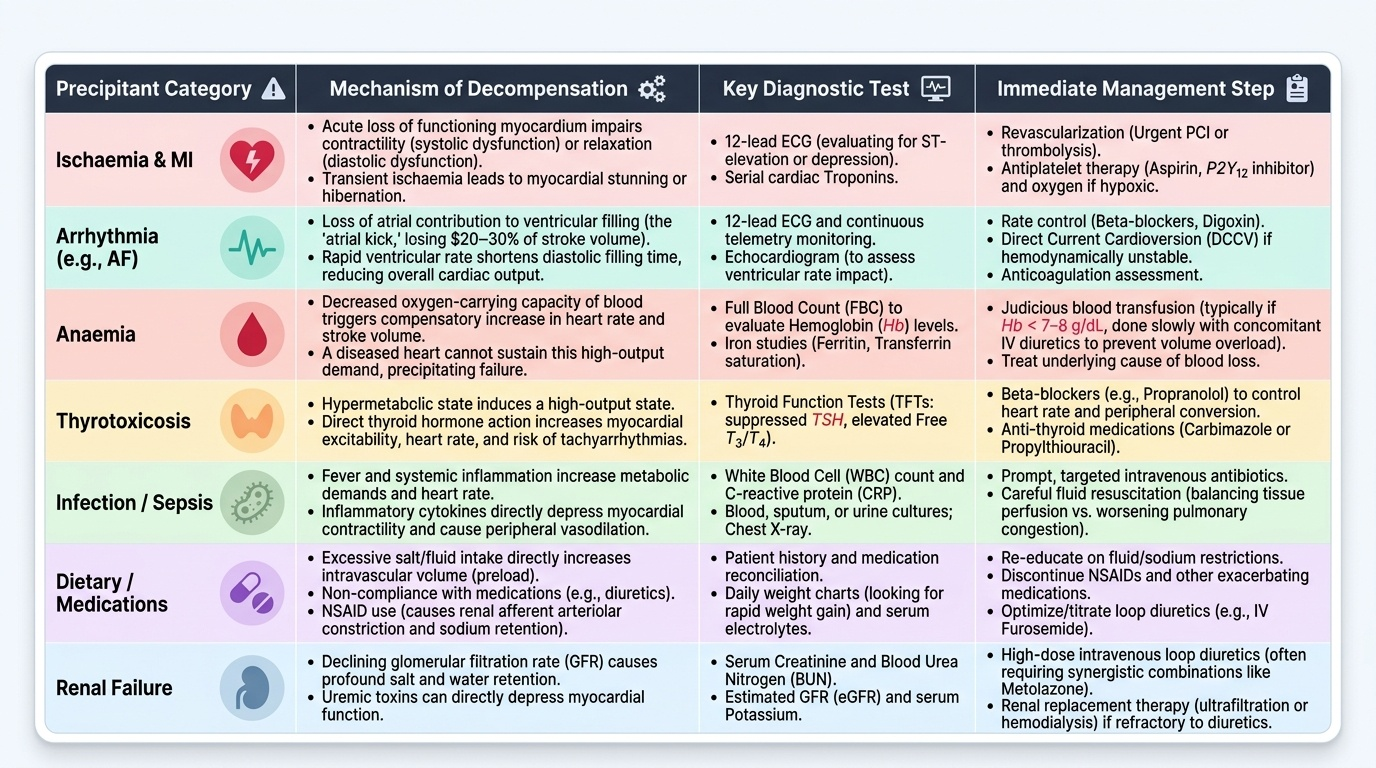
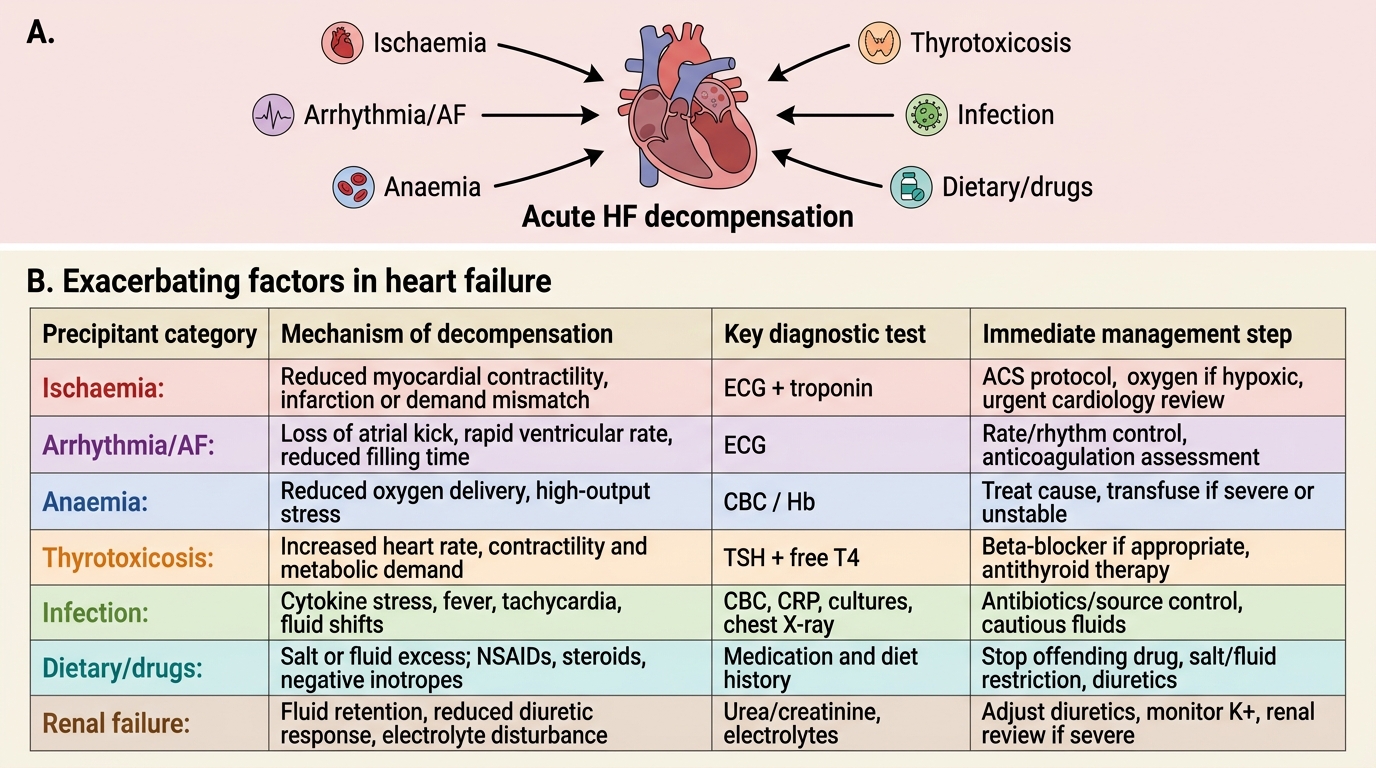
Patients with established, compensated heart failure often present acutely decompensated because of a precipitating factor superimposed on their underlying cardiac dysfunction. Identifying and correcting the precipitant is as important as treating the acute congestion itself — failure to address the cause of decompensation will lead to rapid re-hospitalisation. The mnemonic FAILURE is sometimes used (though clinically, a systematic approach is more important than any mnemonic): Forgot medications/non-compliance, Arrhythmia, Ischaemia/Infection, Lifestyle (salt/fluid excess), Uncontrolled hypertension, Renal failure, Embolism/pulmonary. Here, we examine each exacerbating category in mechanistic detail as required by IM1.6.
Provided image
1. Ischaemia and myocardial infarction: New ischaemic injury to functioning myocardium acutely impairs contractility (systolic dysfunction) or relaxation (diastolic dysfunction), or both. Even transient ischaemia causes myocardial stunning (prolonged dysfunction after a brief ischaemic episode) or hibernation (chronic dysfunction in chronically hypoperfused but viable myocardium). An acute ST-elevation MI causing a large territory of dysfunction is the most dramatic precipitant of acute heart failure and cardiogenic shock.
2. Arrhythmias: Both tachyarrhythmias and bradyarrhythmias can precipitate decompensation. The most clinically important is atrial fibrillation (AF), which decompensates HF through multiple mechanisms: (a) loss of the atrial contribution to ventricular filling (the 'atrial kick' contributes 20–30% of end-diastolic volume, critical in a stiff, non-compliant ventricle); (b) uncontrolled rapid ventricular rate → reduced diastolic filling time → reduced SV; (c) in prolonged rapid AF, tachycardia-induced cardiomyopathy can develop (a reversible dilated cardiomyopathy if rate-controlled). Ventricular tachycardia (VT) similarly impairs cardiac output by reducing diastolic filling time and altering the sequence of ventricular activation. Complete heart block (complete AV dissociation) drops HR to escape rate (30–45 bpm), causing profound reduction in CO.
3. Anaemia: Anaemia reduces the oxygen-carrying capacity of blood. The compensatory response is an increase in cardiac output (via increased HR and, reflexly, increased SV). In a diseased heart that cannot augment output, anaemia imposes a high-output demand on a low-output pump — this mismatch triggers decompensation. Anaemia is extremely common in HF patients in India (particularly iron deficiency, B12/folate deficiency, anaemia of chronic disease, and in women, postpartum blood loss). Even mild anaemia (Hb 9–10 g/dL) can precipitate decompensation in NYHA Class III–IV patients.
4. Thyrotoxicosis: The thyroid hormones T3 and T4 have positive chronotropic and inotropic effects. Thyrotoxicosis increases HR, reduces SVR (peripheral vasodilation), increases metabolic demands, and promotes AF. This combination creates a high-output state that overwhelms a diseased heart. Thyrotoxicosis must be actively screened for in every patient presenting with new-onset AF or unexplained decompensated HF.
5. Infections: Systemic infections — particularly pneumonia, urinary tract infections, and septicaemia — increase metabolic demand and cardiac workload, activate inflammatory cytokines (which are directly myocardially depressant), and promote tachycardia and arrhythmias. Infective endocarditis is a catastrophic cause — acute valve destruction (acute mitral or aortic regurgitation) causes sudden severe HF by volume-overloading an unprepared ventricle.
6. Dietary factors and medication non-compliance: Excess sodium and fluid intake raises preload and precipitates acute pulmonary congestion — the most common cause of repeated HF admissions in chronic patients. Non-compliance with diuretics, ACE inhibitors, or beta-blockers allows RAAS and SNS reactivation. NSAIDs and COX-2 inhibitors cause sodium retention (via prostaglandin antagonism), blunt the efficacy of diuretics and ACE inhibitors, and can precipitate AKI — all of which worsen HF. Calcium channel blockers (especially diltiazem and verapamil) are negatively inotropic and worsen systolic HF. Thiazolidinediones (pioglitazone) cause fluid retention. Certain chemotherapy agents (doxorubicin, trastuzumab, cyclophosphamide) cause myocardial toxicity.
7. Renal failure (cardiorenal syndrome): Renal dysfunction and heart failure are bidirectionally interlinked in the cardiorenal syndrome (CRS). Reduced cardiac output reduces renal perfusion; renal failure causes fluid and sodium retention, electrolyte disturbances, and increased RAAS activation — each worsening HF. Worsening renal function during HF therapy (especially with diuretics and RAAS inhibitors) must be carefully monitored.
8. Pulmonary embolism: Acute PE causes acute right heart failure by suddenly raising pulmonary vascular resistance, obstructing RV outflow. A massive PE can cause acute cor pulmonale, hypotension, and death.
Exacerbating Factors in Heart Failure
Arrhythmias in Heart Failure: Pathogenesis of Atrial Fibrillation and Ventricular Arrhythmias
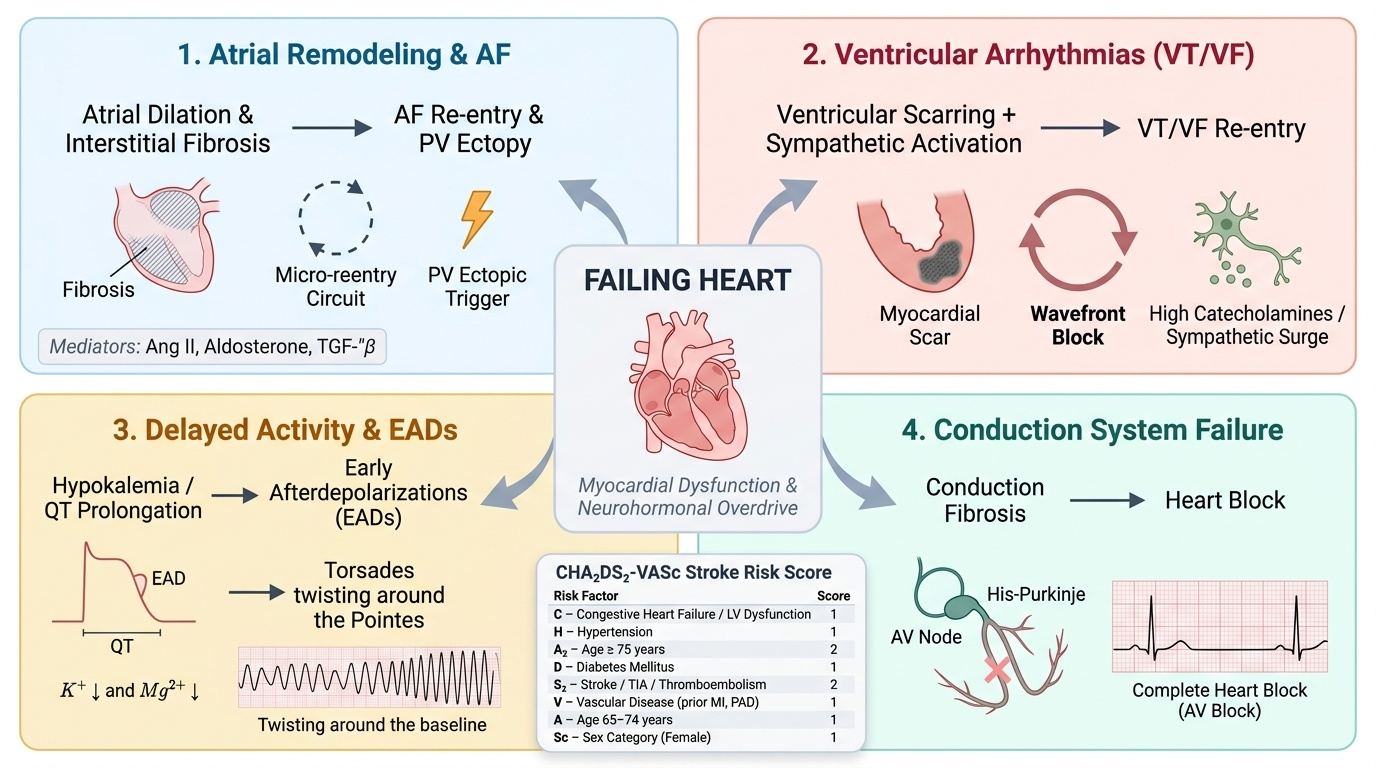
Arrhythmias are both a consequence and a cause of heart failure — a bidirectional relationship that worsens prognosis and complicates management. Atrial fibrillation is present in approximately 30–40% of patients with established heart failure; as HF severity increases (NYHA Class III–IV), the prevalence approaches 50%. At the same time, new-onset AF is a common precipitant of acute decompensation, as described above. Understanding how the failing heart generates arrhythmias requires a mechanistic approach to the three fundamental arrhythmogenic substrates: triggered activity, enhanced automaticity, and re-entry.
Provided image
The structural and neurohormonal milieu of chronic heart failure creates all three substrates simultaneously:
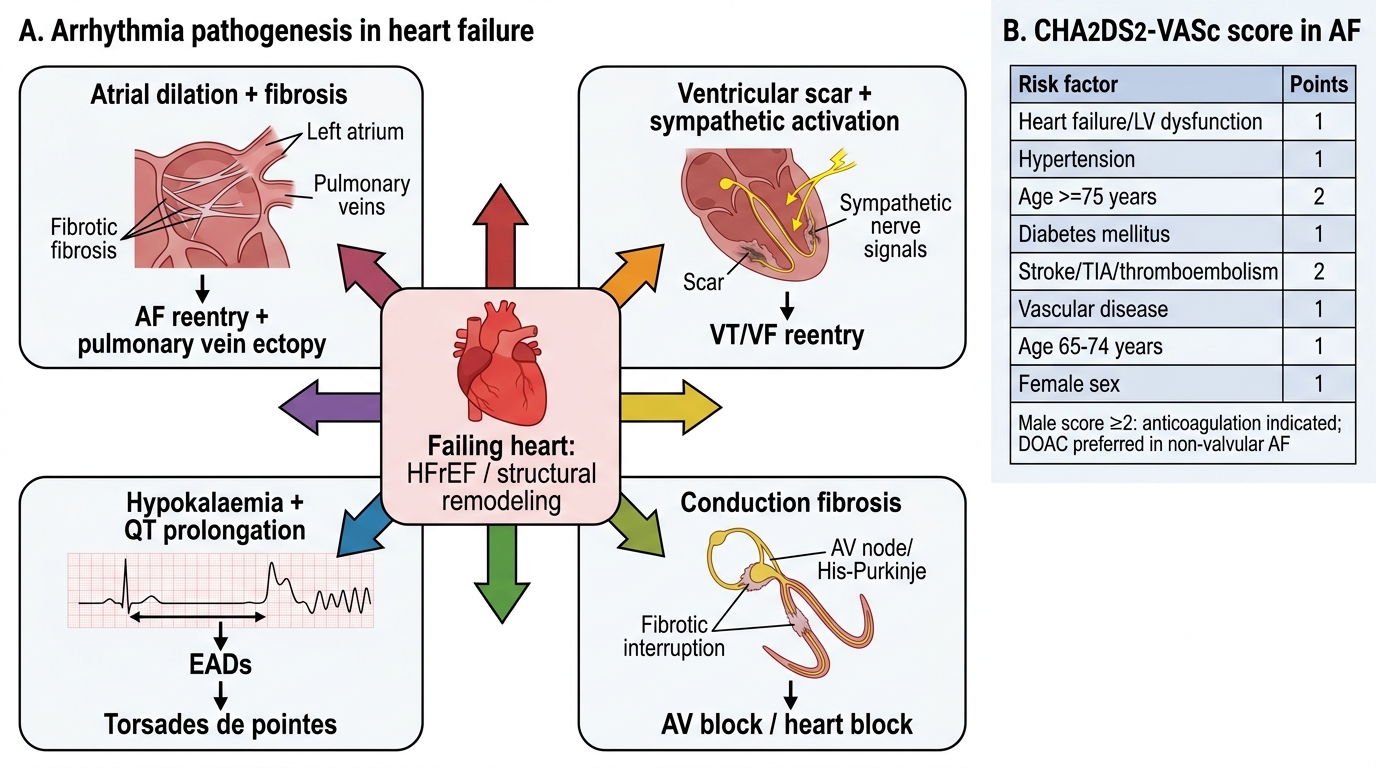
1. Substrate for atrial fibrillation: Chronic atrial pressure and volume overload (from elevated LV filling pressures transmitted backward) causes progressive atrial remodelling — atrial dilation, interstitial fibrosis (mediated by Ang II, aldosterone, and TGF-β), and heterogeneous conduction slowing. Fibrosis creates areas of electrical non-uniformity, setting up micro-reentry circuits that sustain AF. The fibrotic atrium is also less responsive to cardioversion, explaining why AF becomes increasingly persistent as HF progresses. Additionally, sympathetic activation (high catecholamine levels) and electrolyte disturbances (hypokalaemia from diuretic use, hypomagnesaemia) promote triggered ectopic activity from the pulmonary vein ostia — the principal anatomical triggers of AF initiation. Hypokalaemia reduces the resting membrane potential and lengthens the QT interval, promoting early afterdepolarisations (EADs) and triggered activity.
2. Pathogenesis of ventricular arrhythmias (VT/VF): The dilated failing ventricle contains areas of myocardial scar (especially in ischaemic cardiomyopathy) interspersed with hypertrophied but viable muscle. The border zone between scar and viable myocardium is an ideal substrate for macro-reentry circuits underlying monomorphic VT (the most common mechanism in ischaemic HF). In non-ischaemic dilated cardiomyopathy, patchy fibrosis and myocardial disarray (particularly in HCM) create similar reentry substrates. Sympathetic activation raises heart rate and shortens refractory periods, facilitating re-entry. The elevated LVEDP lengthens the QT interval (mechano-electrical coupling) and promotes EADs — the trigger for polymorphic VT (torsades de pointes), especially in the presence of hypokalaemia or QT-prolonging drugs. Sudden cardiac death (SCD) from VF is the leading cause of death in patients with HFrEF (LVEF ≤35%), accounting for 40–50% of deaths — hence the critical role of the implantable cardioverter-defibrillator (ICD) in primary prevention of SCD.
3. Impact of electrolyte disturbances: HF patients receive diuretics (furosemide, hydrochlorothiazide), which cause hypokalaemia and hypomagnesaemia. Both electrolyte disturbances increase arrhythmic risk — hypokalaemia by reducing resting membrane potential (making cells more excitable) and hypomagnesaemia by promoting triggered activity. Careful electrolyte monitoring and replacement are essential components of HF management, not merely supportive care.
Clinical classification of arrhythmias in HF:
| Arrhythmia | Mechanism | Clinical impact in HF |
|---|---|---|
| Atrial fibrillation | Atrial fibrosis + reentry + PV trigger ectopy | Loss of atrial kick, rate-related cardiomyopathy, stroke risk |
| Ventricular tachycardia (VT) | Scar-border reentry (ischaemic); fibrosis reentry (DCM) | Haemodynamic compromise, SCD risk |
| Ventricular fibrillation | Degeneration of VT or primary VF | SCD |
| Ventricular ectopics (PVCs) | EADs, triggered activity | Bigeminy impairs CO; frequent PVCs → cardiomyopathy |
| Complete heart block | Conduction system fibrosis | Severe bradycardia, syncope, worsened CO |
Stroke risk from AF in HF: AF in the context of heart failure confers a particularly high thromboembolic risk. The CHA₂DS₂-VASc score is used to quantify AF-related stroke risk and guide anticoagulation decisions: Congestive heart failure (1 point), Hypertension (1), Age ≥75 (2), Diabetes (1), prior Stroke/TIA (2), Vascular disease (1), Age 65–74 (1), Sex category female (1). A score ≥2 in men or ≥3 in women warrants anticoagulation. Direct oral anticoagulants (DOACs — apixaban, rivaroxaban, dabigatran) are preferred over warfarin in non-valvular AF except in moderate-to-severe mitral stenosis or mechanical heart valves, where warfarin with INR 2–3 (or 2.5–3.5 for mechanical mitral valve) remains standard.
Arrhythmia Pathogenesis in Heart Failure
SELF-CHECK
A 68-year-old man with HFrEF (LVEF 30%) develops rapid atrial fibrillation with a ventricular rate of 140 bpm. He has no mitral stenosis and no mechanical valve. He has the following CHA₂DS₂-VASc features: heart failure (1), hypertension (1), age 68 (1 for age 65–74), diabetes (1), no prior stroke, no vascular disease. His CHA₂DS₂-VASc score is 4. Which anticoagulation is most appropriate?
A. No anticoagulation — score does not meet threshold
B. Aspirin alone
C. Warfarin with target INR 2.5–3.5
D. A direct oral anticoagulant (DOAC) such as apixaban
Reveal Answer
Answer: D. A direct oral anticoagulant (DOAC) such as apixaban
A CHA₂DS₂-VASc score of 4 in a male patient is well above the threshold for anticoagulation (≥2 in men). DOACs (apixaban, rivaroxaban, dabigatran) are the preferred agents in non-valvular AF, offering equivalent or superior efficacy to warfarin with a better safety profile. Warfarin INR 2.5–3.5 is for mechanical mitral valves or severe mitral stenosis — this patient has neither. Warfarin INR 2–3 would be appropriate if a DOAC is contraindicated, but is no longer the first choice in non-valvular AF. Aspirin alone is not recommended for AF stroke prevention. Not anticoagulating a CHA₂DS₂-VASc score of 4 would be a dangerous error.