Page 1 of 23
IM10.1-3 | AKI Foundations — SDL Guide
Learning Objectives
- Define acute kidney injury (AKI) using KDIGO staging criteria for serum creatinine and urine output
- Classify AKI into pre-renal, intrinsic renal, and post-renal categories with their underlying causes
- Describe the pathophysiology of each AKI category, particularly acute tubular necrosis
- Outline the natural history and evolution of AKI from initiation through recovery
- Identify the priorities in initial management of AKI and indications for renal replacement therapy
INSTRUCTIONS
Acute kidney injury is one of the most common and potentially lethal complications encountered in hospitalised patients across all clinical specialties. This module builds your understanding from definition through pathophysiology to management, equipping you to recognise AKI early, categorise its cause, and initiate appropriate priority interventions. The KDIGO framework used throughout is the international standard.
References
- Harrison's Principles of Internal Medicine, 21st ed., Ch. 307 — Acute Kidney Injury (textbook)
- API Textbook of Medicine, 10th ed., Ch. Nephrology — Acute Kidney Injury (textbook)
- KDIGO Clinical Practice Guideline for Acute Kidney Injury, 2012 (guideline)
- Davidson's Principles & Practice of Medicine, 23rd ed., Ch. 17 — Kidney and Urinary Tract Disease (textbook)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
Rajesh, a 55-year-old farmer, is brought to the emergency department with a 3-day history of vomiting and diarrhoea following a feast. He appears sunken-eyed, his mucous membranes are parched, and his skin tents on pinching. His blood pressure is 80/50 mmHg lying down and he is barely producing urine — the catheter bag contains 30 mL over the past 4 hours. His serum creatinine has jumped from a baseline of 0.9 mg/dL (measured 2 months ago at a health camp) to 3.8 mg/dL today. Now consider a second patient: Kavitha, a 40-year-old woman admitted 3 days ago for a pelvic surgery, who received gentamicin for a post-operative fever. Her urine output has been falling despite normal blood pressure and adequate fluid balance. Her creatinine today is 3.1 mg/dL, up from 0.8 mg/dL. Both have acute kidney injury — but the mechanism, reversibility, and treatment approach differ fundamentally. Rajesh's kidneys are starved of blood and will respond rapidly to fluids; Kavitha's tubules are injured by a nephrotoxin and require careful supportive management while they recover. Recognising this distinction within the first hour of evaluation can be the difference between rapid recovery and dialysis.
WHY THIS MATTERS
Acute kidney injury is not a single-specialty concern — it crosses cardiology (cardiorenal syndrome), hepatology (hepatorenal syndrome), surgery (post-operative AKI), intensive care (sepsis-AKI), and general medicine. In India, common causes include gastroenteritis-related dehydration, falciparum malaria, leptospirosis, snake envenomation, and indiscriminate use of NSAIDs and traditional herbal preparations. Hospital-acquired AKI from contrast media and aminoglycosides is increasingly prevalent. For the NMC competencies IM10.1–10.3, you must achieve the KH (Knowledge with clinical application) level — not merely recall definitions, but apply the pre-renal/intrinsic/post-renal framework and the KDIGO staging to a patient encounter and formulate a management priority list.
RECALL
Before proceeding, reactivate your foundation knowledge. The kidney receives approximately 20–25% of cardiac output via the renal arteries, making it exquisitely sensitive to haemodynamic change. Within the renal parenchyma, each nephron consists of a glomerulus (the filtration unit, producing approximately 180 L/day of ultrafiltrate) and a tubule (which reabsorbs about 99% of that filtrate). The glomerular filtration rate (GFR) is the volume of plasma filtered per unit time — normal is approximately 90–120 mL/min/1.73 m². Serum creatinine is an imperfect but widely available surrogate for GFR: it rises as GFR falls, but it lags behind and is affected by muscle mass, diet, and hydration. Recall also that the proximal tubule is metabolically the most active segment, entirely dependent on mitochondrial oxidative phosphorylation and therefore most vulnerable to ischaemia and nephrotoxins — this is why acute tubular necrosis (ATN) predominantly injures the proximal tubule and thick ascending limb.
Defining Acute Kidney Injury — The KDIGO Framework
Acute kidney injury (AKI) is defined by the KDIGO (Kidney Disease: Improving Global Outcomes) 2012 Clinical Practice Guideline as any of the following occurring within 48 hours: an increase in serum creatinine by ≥0.3 mg/dL (≥26.5 µmol/L), a rise in serum creatinine to ≥1.5 times the known or presumed baseline within the prior 7 days, or a urine output of <0.5 mL/kg/h for ≥6 consecutive hours. This operational definition replaced the older, inconsistent terminology of 'acute renal failure' (ARF), 'acute renal insufficiency', and the various RIFLE and AKIN criteria that preceded it. The advantage of the KDIGO definition is precision: it does not require absolute creatinine values (which vary enormously by baseline), but rather detects change — a small rise in an elderly patient with a low baseline can represent a substantial loss of functioning nephron mass.
Provided image
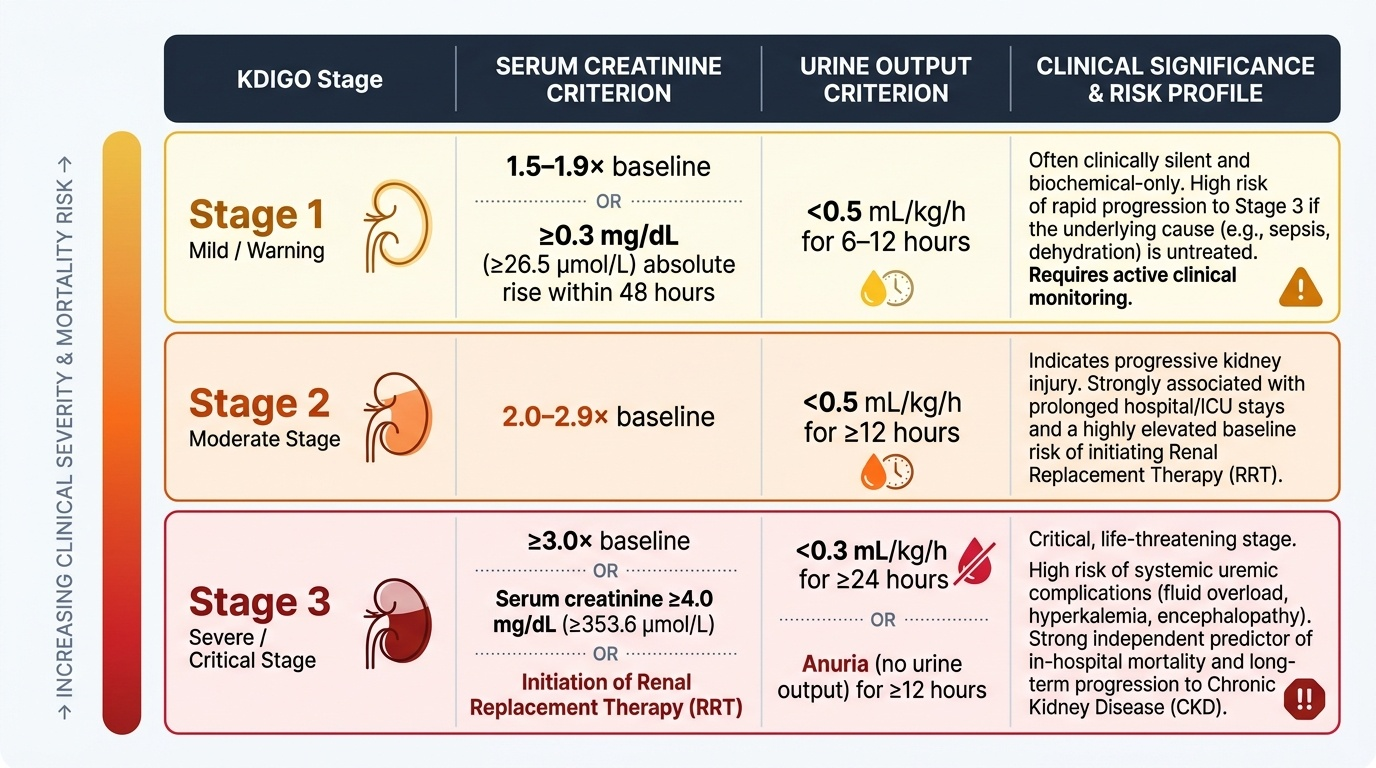
The KDIGO staging system stratifies severity into three stages based on the magnitude of creatinine rise and the degree of oliguria. This staging directly predicts the risk of requiring renal replacement therapy, in-hospital mortality, and long-term risk of progression to chronic kidney disease. Importantly, Stage 1 may appear clinically benign but deserves active attention because progressive injury can advance to Stage 3 within hours if the precipitating cause is not addressed.
| KDIGO Stage | Serum Creatinine Criterion | Urine Output Criterion |
|---|---|---|
| Stage 1 | 1.5–1.9× baseline OR ≥0.3 mg/dL rise within 48 h | <0.5 mL/kg/h for 6–12 h |
| Stage 2 | 2.0–2.9× baseline | <0.5 mL/kg/h for ≥12 h |
| Stage 3 | ≥3.0× baseline OR creatinine ≥4.0 mg/dL OR initiation of RRT | <0.3 mL/kg/h for ≥24 h OR anuria for ≥12 h |
The clinical presentation of AKI spans a wide spectrum. In the early phase, the patient may have no symptoms and the diagnosis is biochemical, caught on routine blood tests in an inpatient with a risk factor (sepsis, nephrotoxin exposure, hypotension). As AKI progresses, oliguria (urine output <400 mL/24 h) or anuria (<100 mL/24 h) becomes the cardinal sign. Systemic manifestations of uraemia — nausea, vomiting, confusion, pericarditis, asterixis — appear as nitrogenous waste accumulates. Fluid overload causes peripheral oedema, pulmonary oedema, and hypertension. Electrolyte disturbances, particularly hyperkalaemia, are life-threatening and must be detected early by ECG and serum potassium monitoring.
Classification: Pre-Renal, Intrinsic Renal, and Post-Renal AKI
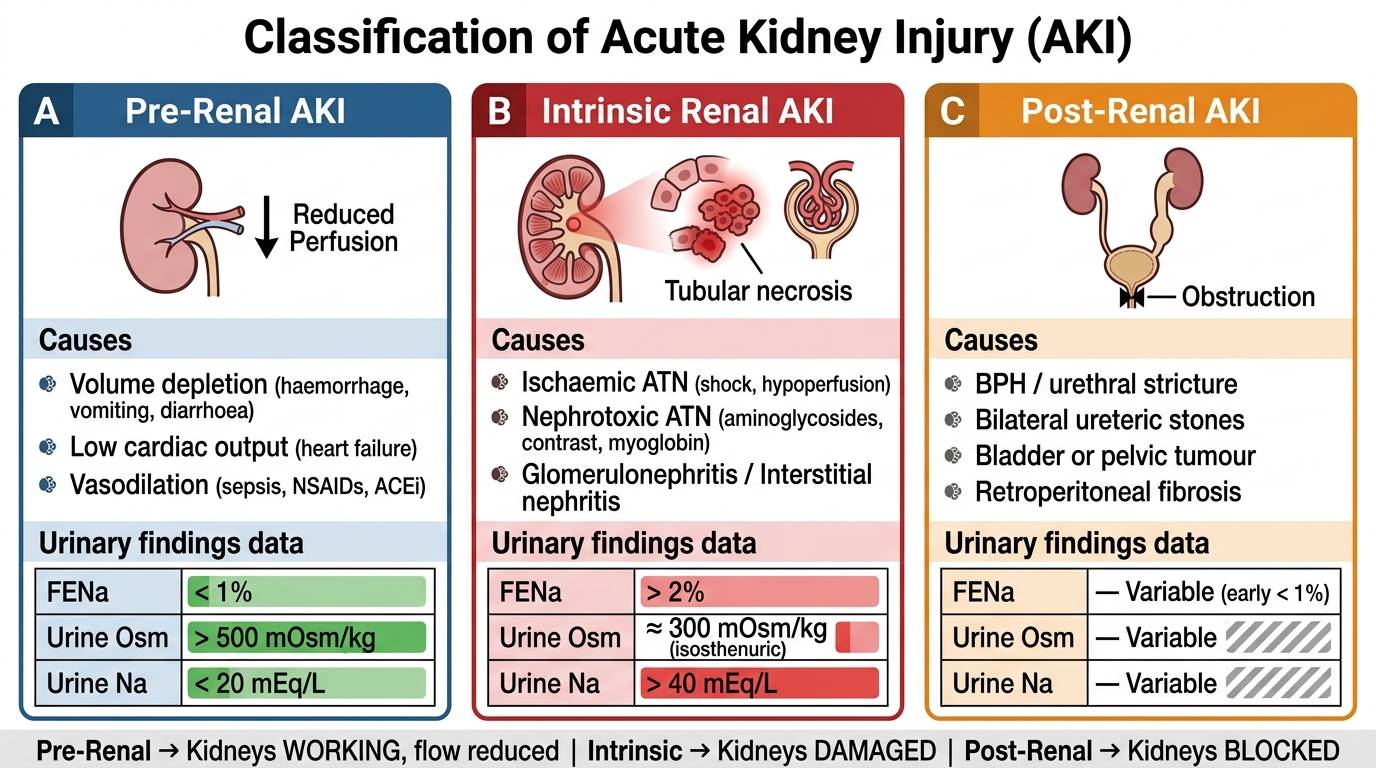
The single most important organising principle in AKI is the pre-renal / intrinsic renal / post-renal classification, which directly determines the diagnostic workup, the urgency of intervention, and the expected trajectory of recovery. This framework asks a deceptively simple question: is the kidney's filtration rate falling because of inadequate blood delivery (pre-renal), because the nephrons themselves are damaged (intrinsic), or because the outflow of urine is obstructed (post-renal)? The answer reshapes every subsequent decision.
Pre-renal AKI accounts for approximately 50–70% of community-acquired AKI. The kidney parenchyma is structurally intact; the glomerular filtration rate has fallen because renal perfusion is inadequate. The kidney compensates by maximally conserving salt and water — producing small volumes of highly concentrated, sodium-poor urine. This is the kidney responding appropriately to what it perceives as volume depletion. The key feature is rapid reversibility: restore the perfusion pressure and GFR recovers within hours. The major categories of pre-renal causes are: (1) true volume depletion — haemorrhage, vomiting, diarrhoea, burns, third-spacing; (2) effective circulating volume depletion despite total body fluid excess — heart failure, hepatic cirrhosis with ascites, nephrotic syndrome; (3) renal artery hypoperfusion — bilateral renal artery stenosis, drugs that reduce glomerular filtration pressure (ACE inhibitors and angiotensin receptor blockers, which reduce efferent arteriolar tone; NSAIDs, which reduce afferent arteriolar dilation by blocking prostaglandins). If pre-renal AKI is not corrected promptly, the ischaemia eventually injures the tubular epithelium and converts pre-renal AKI into ischaemic acute tubular necrosis (ATN) — a much harder condition to reverse. This transition is the clinical turning point.
Intrinsic renal AKI (also called intrinsic or parenchymal AKI) implies structural damage to the kidney itself. The anatomical sub-classification by affected compartment is clinically important:
- Tubular (ATN): the most common intrinsic cause (~70–80% of hospitalised intrinsic AKI). Caused by ischaemia (prolonged pre-renal state, septic shock, major surgery) or nephrotoxins. Key nephrotoxins include aminoglycosides (gentamicin, amikacin), contrast media, cisplatin, amphotericin B, myoglobin (rhabdomyolysis), and haemoglobin (haemolytic conditions).
- Glomerular: acute glomerulonephritis (post-streptococcal GN, IgA nephropathy in acute flare, rapidly progressive GN), characterised by haematuria, proteinuria, hypertension, and the nephritic syndrome. Also includes thrombotic microangiopathy (HUS/TTP) with its triad of MAHA, thrombocytopenia, and organ dysfunction.
- Vascular: renal artery or vein thrombosis, cholesterol emboli, scleroderma renal crisis.
- Interstitial: acute interstitial nephritis (AIN) from drug hypersensitivity (penicillins, cephalosporins, NSAIDs, proton pump inhibitors, rifampicin — important in India for NTEP-treated patients) or infection (leptospirosis, hantavirus). AIN classically presents with fever, rash, eosinophilia, and eosinophiluria — the 'allergic triad', though all three are present together in fewer than 30% of cases.
Post-renal AKI (obstructive uropathy) is caused by obstruction to urine outflow at any level from the collecting system to the urethral meatus. Because the kidney is a paired organ, unilateral obstruction causes AKI only if the contralateral kidney is absent, non-functional, or also obstructed. Causes include: bilateral ureteric obstruction (retroperitoneal fibrosis, bilateral calculi, pelvic tumour invasion), bladder outlet obstruction (benign prostatic hyperplasia — the commonest cause in elderly men in India, urethral stricture, bladder clot retention, neurogenic bladder), and intrinsic ureteric obstruction (bilateral calculi, papillary necrosis — important in diabetics and sickle cell patients). The hallmark is hydronephrosis on ultrasound. Relief of obstruction is followed by a post-obstructive diuresis — a brisk and potentially dangerous polyuria that requires careful fluid replacement to prevent severe dehydration.
Classification of AKI: Pre-Renal vs Intrinsic vs Post-Renal — Aetiology and Urinary Indices
SELF-CHECK
A 70-year-old man with benign prostatic hyperplasia presents with inability to void for 18 hours, lower abdominal pain, and a bladder palpable up to the umbilicus. His serum creatinine is 2.8 mg/dL, up from 1.0 mg/dL 1 month ago. Renal ultrasound shows bilateral hydronephrosis. What is the MOST appropriate immediate intervention?
A. IV normal saline 1 L bolus
B. Furosemide 40 mg IV
C. Urethral catheterisation
D. Urgent haemodialysis
Reveal Answer
Answer: C. Urethral catheterisation
This is post-renal AKI from bladder outlet obstruction (BPH). The correct and immediate intervention is urethral catheterisation to relieve the obstruction. IV saline is inappropriate — fluid overload may worsen with an obstructed bladder. Furosemide will not work if there is outflow obstruction. Dialysis is not indicated when a structural cause can be immediately relieved. After catheterisation, monitor for post-obstructive diuresis and replace fluid losses cautiously.
Pathophysiology of Acute Tubular Necrosis
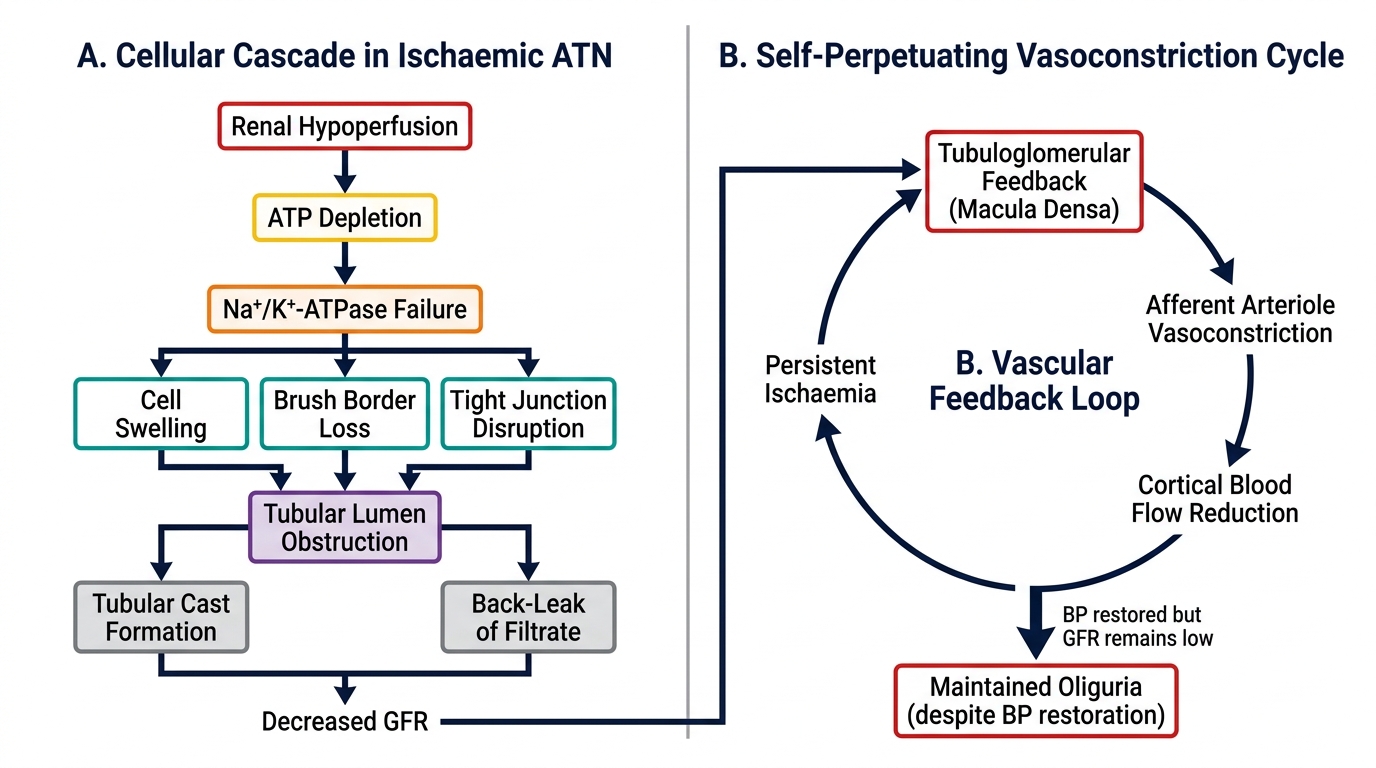
Acute tubular necrosis (ATN) is the most common cause of intrinsic AKI in hospitalised patients and deserves detailed understanding because its pathophysiology explains both the clinical phases and the therapeutic limitations. The tubular epithelial cell — particularly in the proximal tubule (S3 segment) and the medullary thick ascending limb — is the primary target. These segments are uniquely vulnerable because they combine high metabolic activity (requiring abundant ATP for active transport) with borderline oxygen delivery (the renal medulla operates at the edge of hypoxia even under normal conditions, since the descending vasa recta deliver oxygen that is already partially shunted to the ascending vasa recta by countercurrent exchange).
In ischaemic ATN, the sequence of injury proceeds as follows. Renal hypoperfusion depletes intracellular ATP in tubular cells within minutes. ATP depletion disrupts the Na/K-ATPase on the basolateral membrane, causing intracellular sodium accumulation, cellular oedema, and loss of the normal brush border polarity. The tight junctions between tubular cells loosen, allowing backdiffusion of glomerular filtrate across the tubular wall into the peritubular capillaries — this is a key mechanism by which the apparent GFR falls disproportionately to the actual nephron loss. Cellular oedema and detachment of dead tubular cells causes tubular cast formation, and these casts obstruct the lumens, further raising intratubular pressure and suppressing filtration. Additionally, sustained cortical vasoconstriction (mediated by endothelin and reduced nitric oxide) perpetuates medullary ischaemia even after systemic blood pressure is restored — explaining why GFR does not immediately recover with volume resuscitation once ATN is established.
In nephrotoxic ATN, the mechanism varies by agent. Aminoglycosides accumulate in the proximal tubular cells by endocytosis via megalin, generating reactive oxygen species (ROS) that damage mitochondria. They also cause direct tubulotoxicity to the apical membrane. Contrast media cause vasoconstriction of the afferent arteriole (mediated by adenosine and endothelin), reduce medullary blood flow, generate ROS, and may cause direct tubulotoxic injury — the combination creates contrast-induced AKI (CI-AKI). In rhabdomyolysis, myoglobin released from crushed muscle is filtered at the glomerulus; in acidic urine it precipitates as ferrihemate casts, causing tubular obstruction, and its iron moiety generates ROS that directly injure tubular epithelium. Volume depletion and aciduria (pH <6) dramatically amplify myoglobin-mediated toxicity — hence aggressive alkaline hydration is the cornerstone of rhabdomyolysis management.
Pathophysiology of Ischaemic Acute Tubular Necrosis (ATN)