Page 1 of 17
IM12.{1-3,11} | Thyroid Dysfunction Foundations — SDL Guide
Learning Objectives
- Describe the epidemiology and public health burden of hypothyroidism, hyperthyroidism, and iodine deficiency disorders in India
- Explain the physiology of the hypothalamic-pituitary-thyroid axis including hormone synthesis, transport, and feedback
- Discuss the pathogenesis of Hashimoto thyroiditis and Graves disease, including the role of autoimmunity
- Describe the principle, indications, and interpretation of radioactive iodine uptake (RAIU) in thyroid diagnosis
- Describe the National Iodine Deficiency Disorders Control Programme (NIDDCP), its objectives, interventions, and monitoring indicators
INSTRUCTIONS
This module covers the foundational science of thyroid dysfunction — epidemiology, HPT axis physiology, pathogenesis of autoimmune and iodine-deficiency thyroid disease, radioiodine uptake principles, and India's public health response to iodine deficiency. It maps to NMC competencies IM12.1, IM12.2, IM12.3, and IM12.11 at the Knowledge and KH levels.
References
- Harrison's Principles of Internal Medicine, 21st ed., Ch. 400 — Disorders of the Thyroid Gland (textbook)
- API Textbook of Medicine, 10th ed. — Thyroid Disorders (textbook)
- Davidson's Principles and Practice of Medicine, 24th ed. — Thyroid Disease (textbook)
- National Iodine Deficiency Disorders Control Programme, Government of India — Programme Guidelines (guideline)
- Indian Thyroid Society Guidelines for Diagnosis and Management of Thyroid Disorders, 2019 (guideline)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
In the outpatient department, two patients sit in adjacent chairs. The first is a 32-year-old woman referred for weight loss, palpitations, and tremor — she has lost 8 kg in 3 months despite eating more than ever, and her resting heart rate is 112 beats per minute. The second is a 48-year-old woman who was sent by her obstetrician after her second miscarriage; she reports fatigue, weight gain, constipation, and a slowed voice, and her resting heart rate is 54 beats per minute. On examining their necks you find, in the first patient, a soft diffuse goitre with an audible bruit; in the second, a firm, rubbery, nodular goitre. Their thyroid function tests arrive: Patient 1 has undetectable TSH with elevated Free T4 and Free T3; Patient 2 has elevated TSH with a low Free T4. These are mirror images of each other — both arising from the same gland, driven by fundamentally opposite mechanisms: one by autoimmune stimulation, the other by autoimmune destruction. Understanding thyroid disease means understanding its epidemiology, the physiology of the axis that controls it, the role of iodine and immunity in its pathogenesis, and the rationale of radioiodine as a diagnostic and therapeutic tool.
WHY THIS MATTERS
Thyroid disease is one of the most prevalent endocrine disorders in India, with an estimated 42 million people affected. Hypothyroidism alone affects approximately 2–3% of women. India carries a massive burden of iodine-deficiency disorders (IDD), with 263 million people at risk in endemic zones — a burden the government has addressed through universal salt iodisation under the National Iodine Deficiency Disorders Control Programme (NIDDCP). As a final-year student in General Medicine, you must be able to describe the epidemiology, pathogenesis, and physiological basis of thyroid dysfunction (IM12.1, IM12.2), explain the role of radioiodine uptake in diagnosis (IM12.3), and discuss India's iodisation programme (IM12.11). These form the foundational knowledge that underpins clinical assessment, investigation ordering, and management decisions covered in subsequent modules.
RECALL
Before proceeding, activate what you already know. The thyroid gland is a bilobed endocrine gland in the anterior neck, weighing approximately 20–25 g in adults. It synthesises two iodine-containing hormones: thyroxine (T4) and triiodothyronine (T3), the biologically active form. Thyroid synthesis requires iodine, an essential trace element obtained exclusively through diet. The gland is regulated by thyroid-stimulating hormone (TSH) from the anterior pituitary, which is itself regulated by thyrotrophin-releasing hormone (TRH) from the hypothalamus. Negative feedback from circulating T4/T3 suppresses both TSH and TRH. Recall also the key autoimmune diseases from your second-year immunology: Graves disease involves stimulatory antibodies; Hashimoto thyroiditis involves destructive lymphocytic infiltration. These fundamentals now need to be deepened with epidemiological data, molecular pathogenesis, and the principles of radioiodine uptake.
Epidemiology and Public Health Burden of Thyroid Disease
Thyroid disease presents a significant and often underestimated public health burden in India, shaped by both nutritional deficiencies and the rising tide of autoimmune disease. Understanding the epidemiology is not merely academic — it informs clinical suspicion, screening policy, and public health intervention, all of which the NMC expects you to apply in practice.
Hypothyroidism affects approximately 2–3% of the general population globally, with prevalence rising steeply in women (particularly after the fourth decade) and in populations with iodine deficiency or autoimmune predisposition. In India, data from the Indian Thyroid Society (ITS) and national surveys estimate that 1 in 10 Indians suffers from some form of thyroid disorder, with hypothyroidism dominating. Subclinical hypothyroidism — characterised by elevated TSH with normal free T4 — is even more prevalent, affecting up to 8–10% of women over 40. Overt hypothyroidism is more common in women (female-to-male ratio approximately 6–8:1), reflecting the predominance of autoimmune thyroiditis (Hashimoto thyroiditis) as its primary cause in iodine-sufficient populations.
Hyperthyroidism (thyrotoxicosis) affects approximately 0.5–1% of the population, with Graves disease accounting for 70–80% of cases in iodine-sufficient regions. The female preponderance is even more pronounced (8:1). Toxic multinodular goitre and toxic adenoma become relatively more important in iodine-deficient areas and in older patients. In contrast to the insidious onset of Graves disease in younger women, toxic multinodular goitre tends to affect older individuals and presents with a more gradual clinical course.
Iodine deficiency disorders (IDD) represent a unique dimension of Indian thyroid epidemiology. Iodine is not uniformly distributed in soil and water, and in mountainous, sub-Himalayan, and inland regions, the natural dietary supply is insufficient. Approximately 263 million Indians live in iodine-deficient areas. IDD encompasses a spectrum from simple diffuse goitre (the most visible manifestation) to hypothyroidism, cretinism (in severe congenital deficiency), impaired cognitive development in children, and increased risk of thyroid cancer (follicular carcinoma is more common in iodine-deficient populations). The most devastating consequence — endemic cretinism — results from severe iodine deficiency during pregnancy, causing irreversible intellectual disability in the offspring.
Genetic predisposition plays a meaningful role in autoimmune thyroid diseases: HLA-DR3 is associated with Graves disease; HLA-DR4 and DR5 with Hashimoto thyroiditis. Family history of autoimmune thyroid disease, type 1 diabetes, or other organ-specific autoimmune conditions significantly increases an individual's risk. However, genetic factors alone do not determine disease expression — environmental triggers (iodine excess, infections, smoking, stress, postpartum state) are required co-factors.
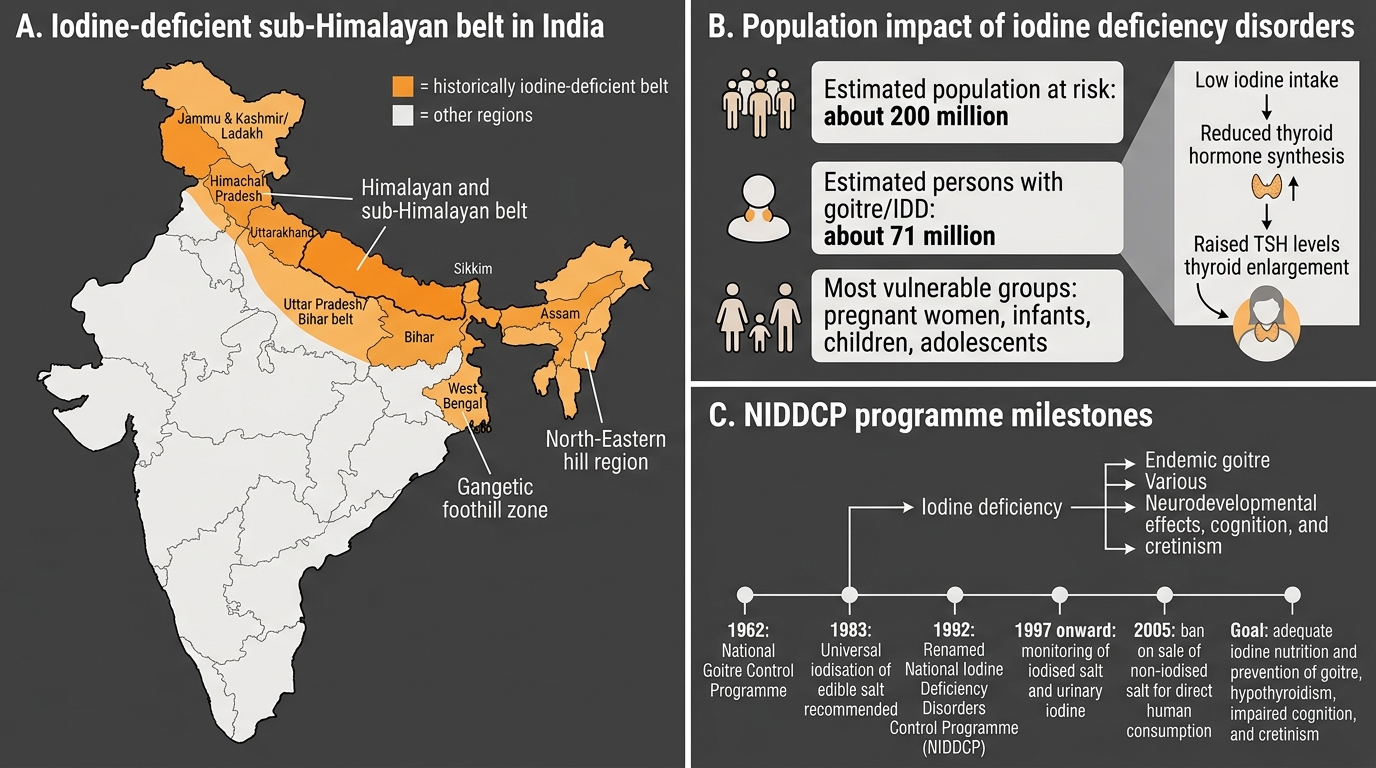
Iodine Deficiency Disorders and NIDDCP in India
Physiology of the Hypothalamic-Pituitary-Thyroid Axis
The hypothalamic-pituitary-thyroid (HPT) axis is the regulatory hierarchy that maintains thyroid hormone levels within a narrow physiological range. A thorough understanding of this axis — including its feedback loops, the biochemistry of thyroid hormone synthesis, and the mechanisms of peripheral action — is essential for interpreting thyroid function tests correctly and understanding how disease at each level produces distinct hormonal patterns.
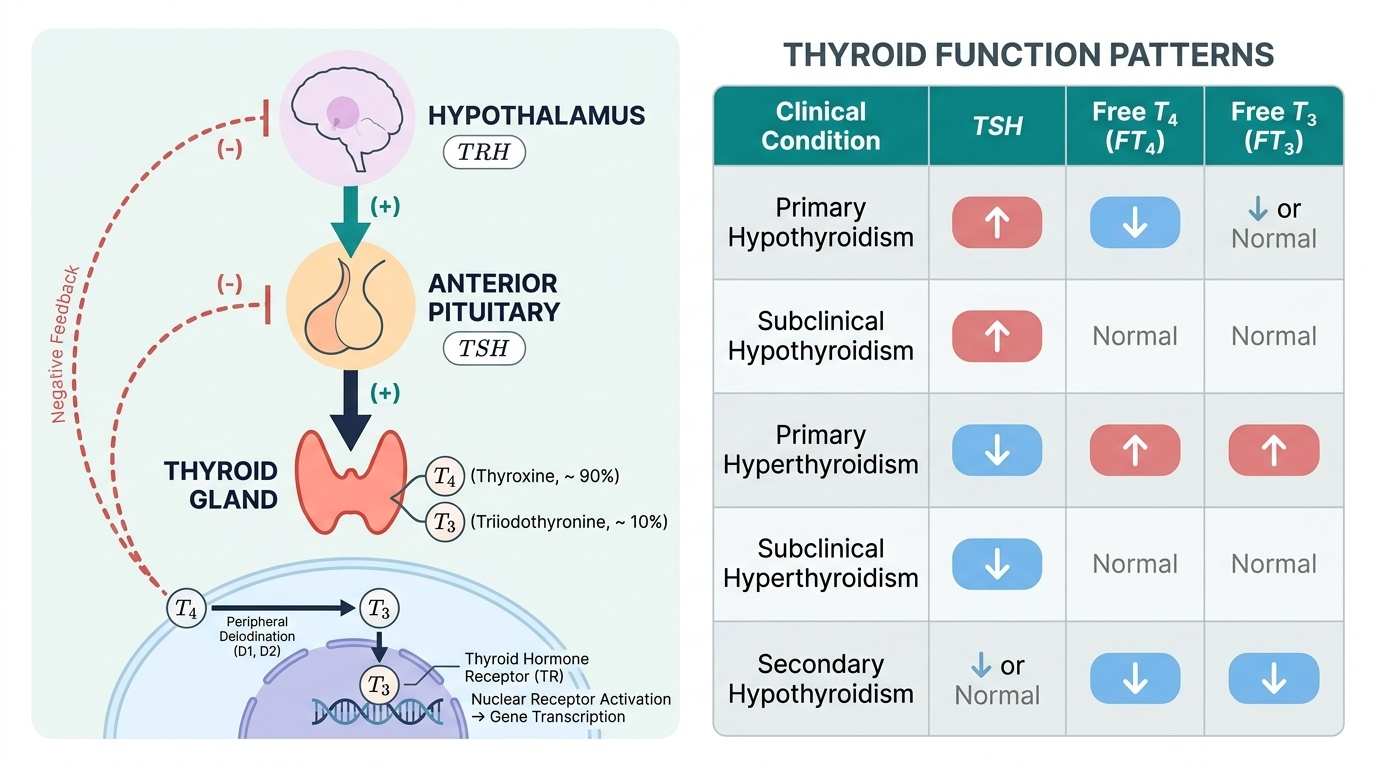
Provided image
Axis structure and feedback: The hypothalamus secretes thyrotrophin-releasing hormone (TRH) from the paraventricular nucleus in a pulsatile, circadian fashion. TRH travels via the hypothalamic-portal system to the anterior pituitary, where it binds thyrotroph cells and stimulates release of thyroid-stimulating hormone (TSH). TSH acts on the thyroid gland through the TSH receptor (TSHR), a G-protein coupled receptor that activates adenylyl cyclase. The resulting cAMP cascade drives all steps of thyroid hormone synthesis: iodide uptake, organification, coupling, and hormone secretion. Circulating T4 and T3 exert negative feedback on both the hypothalamus (suppressing TRH) and the anterior pituitary (suppressing TSH). This negative feedback is the physiological basis of thyroid function testing: in primary hypothyroidism (thyroid failure), falling T4 lifts the feedback brake, and TSH rises; in primary hyperthyroidism, rising T4 intensifies feedback and suppresses TSH.
Thyroid hormone synthesis: The follicular cells of the thyroid synthesise and secrete thyroid hormones through a coordinated sequence. Iodide (I⁻) is actively transported into the follicular cell from the bloodstream by the sodium-iodide symporter (NIS), a basal membrane transporter energised by the Na⁺/K⁺-ATPase gradient. Once inside, iodide is oxidised to reactive iodine by thyroid peroxidase (TPO) (using H₂O₂ generated by DUOX2). Reactive iodine is then incorporated into tyrosine residues of thyroglobulin (Tg), a large glycoprotein stored in the follicular colloid — a process called organification. Monoiodotyrosine (MIT) and diiodotyrosine (DIT) residues are then coupled by TPO: DIT + DIT → T4 (thyroxine) and MIT + DIT → T3 (triiodothyronine). Proteolysis of stored thyroglobulin releases T4 and T3 into the bloodstream. Approximately 90% of thyroid secretion is T4; most circulating T3 arises from peripheral deiodination of T4 by deiodinase enzymes (type I deiodinase in liver and kidney; type II in brain and pituitary). The TPO enzyme and NIS are the targets of antibodies in autoimmune thyroid disease: anti-TPO antibodies (anti-thyroid peroxidase) are the marker of Hashimoto thyroiditis, and are also elevated in Graves disease.
Thyroid hormones in the circulation: T4 and T3 are almost entirely protein-bound in plasma — to thyroxine-binding globulin (TBG) (~70%), prealbumin (transthyretin), and albumin. Only the free (unbound) fraction is biologically active — Free T4 (FT4) and Free T3 (FT3). This has important clinical implications: total T4 levels change with TBG status (oestrogen excess in pregnancy or OCP use raises TBG, raising total T4 but not free T4); measuring free fractions avoids this confound.
Physiological role of T3/T4: Thyroid hormones exert their effects primarily through nuclear thyroid hormone receptors (TR-α and TR-β), which regulate gene transcription. Their actions are wide-ranging: increased basal metabolic rate (via Na⁺/K⁺-ATPase upregulation), increased cardiac rate and contractility, normal development of the brain (critical in neonatal period — congenital hypothyroidism impairs neuronal migration and myelination), linear growth (synergy with growth hormone), gut motility, and skin and hair maintenance. The broad physiological role explains the diverse clinical manifestations of both excess and deficiency.
Pathogenesis of Hypothyroidism and Hyperthyroidism
Understanding the pathogenesis of thyroid dysfunction requires appreciating how autoimmunity, iodine metabolism, and genetic predisposition converge to produce either thyroid destruction (hypothyroidism) or thyroid overstimulation (hyperthyroidism). These are not simply opposite poles of the same spectrum — they involve distinct immunological mechanisms that must be understood separately.
Hashimoto thyroiditis (autoimmune hypothyroidism) is the most common cause of hypothyroidism in iodine-sufficient populations worldwide and the leading cause in India outside of iodine deficiency zones. The pathogenesis centres on CD8+ cytotoxic T lymphocyte-mediated destruction of thyroid follicular cells, accompanied by a TH1-polarised CD4+ T helper response and B-cell production of thyroid-specific autoantibodies. The key autoantibodies are: anti-TPO antibodies (anti-thyroid peroxidase; present in >90% of Hashimoto cases — the most sensitive marker) and anti-thyroglobulin (anti-Tg) antibodies (less specific, present in ~60%). These antibodies contribute to complement-mediated cytotoxicity and facilitate antibody-dependent cellular cytotoxicity (ADCC), but they are also markers of autoimmune activation rather than solely pathogenic. The histological hallmark is lymphocytic infiltration with formation of germinal centres within the thyroid parenchyma, progressive follicular destruction, and eventual fibrosis — giving the gland its characteristic firm, rubbery texture. The disease evolves through phases: initially the autoimmune process may cause transient hyperthyroidism (Hashitoxicosis) as stored hormone leaks from damaged follicles, before progressive hypothyroidism supervenes. Genetic associations include HLA-DR4, DR5, and CTLA4 polymorphisms (impairing T regulatory cell function).
Graves disease (autoimmune hyperthyroidism) is a type II hypersensitivity reaction in which stimulatory TSH receptor antibodies (TRAb) — specifically thyroid-stimulating immunoglobulins (TSI) — bind to and constitutively activate the TSH receptor on follicular cells. Because TSI mimics TSH action but is not subject to normal feedback suppression, the thyroid is continuously driven to produce excess T4 and T3, independent of pituitary control. This is the key mechanistic distinction: in Graves disease, the pituitary response is entirely appropriate — it suppresses TSH to near-zero levels in response to excess T4/T3 — but the driver of hormone excess is the IgG antibody, which the pituitary cannot suppress. The result is autonomous, feedback-resistant overproduction. Clinically, TRAb drives all three features of Graves disease: (1) thyrotoxicosis (metabolic effects of excess T4/T3); (2) diffuse goitre (TSH-receptor stimulation drives gland hypertrophy); and (3) Graves ophthalmopathy (the TSH receptor is also expressed on orbital fibroblasts — TRAb activates these, causing glycosaminoglycan deposition, orbital inflammation, and proptosis). The dermopathy (pretibial myxoedema) and acropachy (finger clubbing-like periosteal change) are less common but pathognomonic. Genetic associations include HLA-DR3 and CTLA4 polymorphisms.
Iodine deficiency as a cause of goitre and hypothyroidism: In iodine-deficient regions, the thyroid cannot synthesise adequate T4/T3, so the pituitary responds by raising TSH levels. Chronically elevated TSH drives follicular cell proliferation and gland hypertrophy, producing endemic goitre — the most visible manifestation of IDD. If iodine deficiency is mild, the gland can partially compensate; if severe, frank hypothyroidism results. During pregnancy, iodine requirements increase substantially, making pregnant women particularly vulnerable — severe maternal hypothyroidism during the first trimester, when fetal thyroid is not yet functional, causes irreversible damage to fetal brain development (endemic cretinism: intellectual disability, deaf-mutism, spasticity). The correction of iodine deficiency through universal salt iodisation is one of the most cost-effective public health interventions in history. Paradoxically, very high iodine intake causes transient thyroid inhibition (Wolff-Chaikoff effect) and may precipitate hypothyroidism in susceptible individuals (those with subclinical Hashimoto thyroiditis, for example).
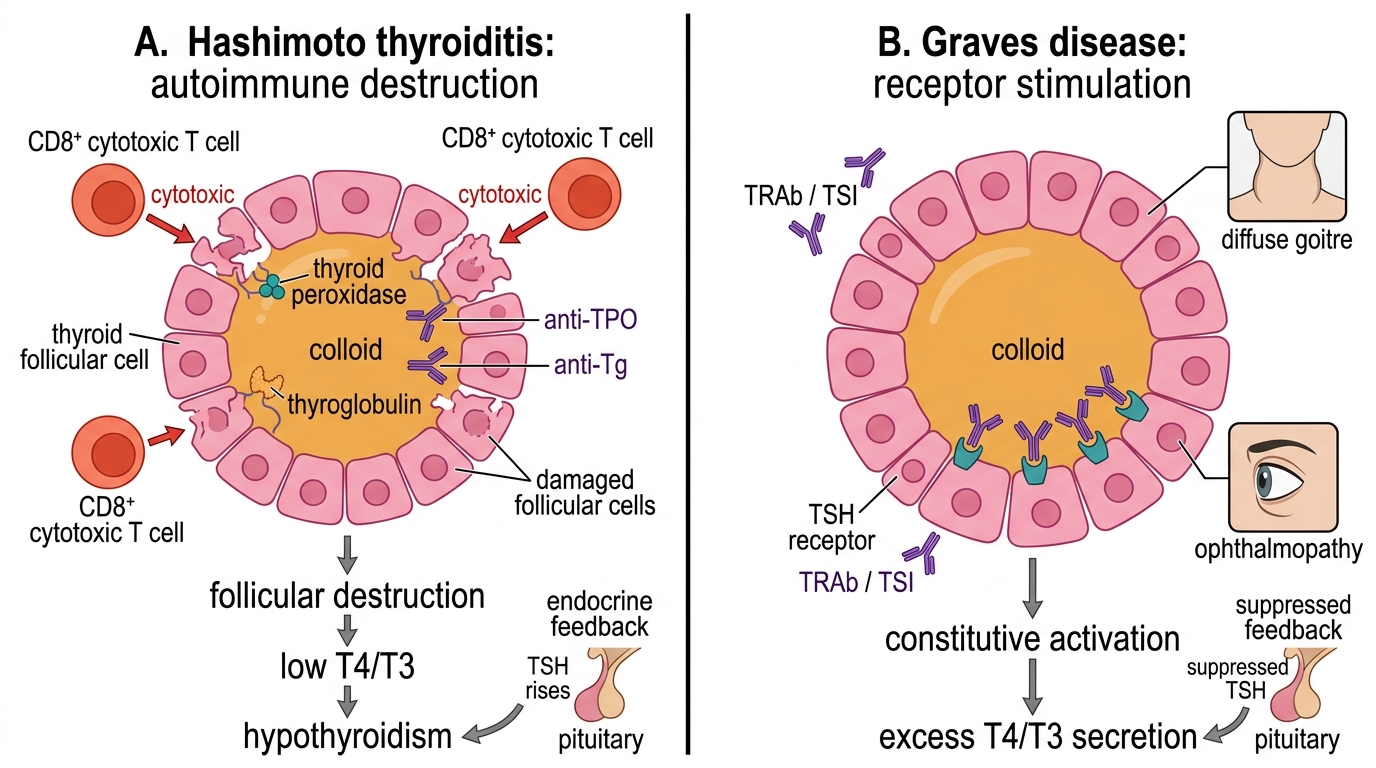
Autoimmune Thyroid Dysfunction: Hashimoto vs Graves
SELF-CHECK
A 35-year-old woman is diagnosed with hyperthyroidism. Her serum TSH is undetectable, Free T4 is markedly elevated, and she has a diffuse goitre with an audible bruit. Serum TSH receptor antibodies (TRAb) are strongly positive. What is the precise mechanism by which her thyroid is overactive despite undetectable TSH?
A. Anti-TPO antibodies destroy the negative feedback loop, preventing TSH suppression
B. Constitutive activating mutation of the TSH receptor in all follicular cells
C. TSH receptor antibodies (TSI) bind and activate the TSH receptor independently of TSH, producing autonomous T4/T3 secretion that is not subject to feedback regulation
D. TRH from the hypothalamus is overproduced, directly stimulating the thyroid without TSH
Reveal Answer
Answer: C. TSH receptor antibodies (TSI) bind and activate the TSH receptor independently of TSH, producing autonomous T4/T3 secretion that is not subject to feedback regulation
In Graves disease, thyroid-stimulating immunoglobulins (TSI/TRAb) mimic TSH by binding to and constitutively activating the TSH receptor on follicular cells. This drives continuous T4/T3 production independent of pituitary control. The pituitary responds appropriately — it suppresses TSH to undetectable levels in response to high T4/T3 — but the antibody driver cannot be switched off by negative feedback, resulting in autonomous hyperthyroidism. Option B describes toxic adenoma (a somatic mutation affecting one nodule, not all cells). Anti-TPO antibodies are markers of autoimmunity but do not directly drive hyperthyroidism in Graves disease.