Page 7 of 14
IM19.7-8 | Movement Disorder Investigation and Parkinson Therapy — SDL Guide
Learning Objectives
- Choose and interpret appropriate diagnostic imaging tests in the evaluation of movement disorders
- Describe the pharmacology, dose, side effects, and drug interactions of the major agents used in Parkinson's disease management
- Discuss the levodopa-carbidopa combination, its long-term motor complications, and strategies to minimise them
- Explain the pharmacological rationale for dopamine agonists, MAO-B inhibitors, COMT inhibitors, anticholinergics, and amantadine in parkinsonism
- Identify dangerous drug interactions relevant to antiparkinson therapy
INSTRUCTIONS
Having established the clinical diagnosis of a movement disorder through history and examination, the clinician must now select, interpret, and apply investigations to confirm the diagnosis, exclude reversible causes, and guide treatment. Simultaneously, for patients with Parkinson's disease — the commonest movement disorder requiring pharmacotherapy — the clinician must understand the pharmacology of the antiparkinson drugs in sufficient detail to initiate therapy, anticipate complications, and counsel patients about long-term drug management. This module covers both these competency areas: IM19.7 (imaging in movement disorders) and IM19.8 (pharmacology of antiparkinson drugs).
References
- Harrison's Principles of Internal Medicine, 21st ed., Ch. 431 — Parkinson's Disease, Management (textbook)
- API Textbook of Medicine, 10th ed., Ch. — Parkinson's Disease: Pharmacotherapy (textbook)
- Movement Disorder Society (MDS) Evidence-Based Medicine Review: Treatments for Parkinson's Disease, 2018 update (guideline)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A 66-year-old retired engineer, Mr Venkataraman, presents with a 3-year history of asymmetric rest tremor, cogwheel rigidity, and bradykinesia. His neurologist is confident of the clinical diagnosis of Parkinson's disease. Mr Venkataraman's son, an engineer himself, has read online that a 'brain scan' can confirm the diagnosis and asks whether a CT or MRI is needed. Meanwhile, a different patient — Mr Ibrahim, 55 years old, recently started on haloperidol for late-onset psychosis — has developed symmetric rest tremor and bradykinesia. His psychiatrist is uncertain whether he has developed drug-induced parkinsonism or has incidentally been found to have idiopathic PD that his antipsychotic unmasked. The neurologist advises a specific nuclear medicine scan to distinguish the two. What are these scans? When are they indicated? And once the diagnosis is confirmed, what drugs will Mr Venkataraman take for the rest of his life — what do they do, how do you dose them, and what are the long-term complications that his family needs to understand?
WHY THIS MATTERS
Diagnostic imaging in movement disorders is frequently misused — CT and MRI are ordered routinely when they add little in typical idiopathic PD, while the specific and diagnostically discriminating nuclear medicine investigations (DAT-SPECT) are underutilised or unknown to many generalists. Understanding when each modality is indicated reduces both cost and diagnostic delay. The pharmacology of antiparkinson drugs is clinically critical because Parkinson's disease is one of the most drug-intensive chronic neurological conditions: patients typically take 3–5 drugs simultaneously, drug interactions are clinically significant (notably with MAO inhibitors and serotonergic drugs), and long-term levodopa complications — wearing-off, dyskinesias, on-off fluctuations — are the central management challenge in advanced disease. The NMC IM19.7 (SH: choose and interpret imaging) and IM19.8 (KH: describe pharmacology, dose, side effects, interactions) competencies reflect the direct patient-care relevance of this material.
RECALL
Before proceeding, recall the key pharmacological principle underlying Parkinson's disease treatment: PD is primarily a disease of dopamine deficiency in the striatum, caused by loss of dopaminergic neurons in the substantia nigra pars compacta. Restoring striatal dopamine — or mimicking its effects — is the basis of virtually all current pharmacotherapy. Recall also that dopamine itself cannot cross the blood-brain barrier (BBB), which is why levodopa (the metabolic precursor) is used — it is actively transported into the brain by the large neutral amino acid transporter and then decarboxylated to dopamine. Also recall from the earlier module that the direct pathway (D1 receptor, pro-movement) and indirect pathway (D2 receptor, anti-movement) are the two arms of basal ganglia circuitry; dopamine facilitates movement by simultaneously activating D1 and inhibiting D2. Drugs that act on D1/D2 receptors, or that increase synaptic dopamine, form the core of antiparkinson pharmacotherapy.
Clinical Presentation: When and Why to Investigate
Parkinson's disease is fundamentally a clinical diagnosis. The clinical criteria (International Parkinson and Movement Disorder Society criteria, 2015) require bradykinesia plus at least one of rest tremor or rigidity, after exclusion of alternative causes. When the clinical picture is typical — asymmetric onset, bradykinesia, rest tremor, cogwheel rigidity, good levodopa response, absence of red flags — no imaging investigation is required to make or confirm the diagnosis. This is an important principle to internalise: the majority of patients with typical idiopathic PD seen in a neurology clinic do not need a DAT-SPECT or MRI as part of their initial diagnostic work-up. The diagnosis is clinical, built on the three-part formulation of anatomical localisation, nature of process, and probable cause, supported by the characteristic history, examination findings, and clinical evolution.
However, there are specific clinical scenarios in which diagnostic imaging is indicated and changes management. The clinician must be able to identify these scenarios and select the appropriate modality — choosing between structural (CT, MRI) and functional/nuclear medicine (DAT-SPECT, MIBG) investigations based on the clinical question being asked. This decision matrix is what NMC competency IM19.7 tests at the SH level: not merely knowing that these scans exist, but understanding when and why to order them, and what a given result means for the differential diagnosis and treatment plan.
Scenario 1 — Diagnostic uncertainty: When the clinical picture is atypical or ambiguous (e.g., pure postural tremor without bradykinesia that could be essential tremor or early PD; or parkinsonism developing rapidly in a young patient where Wilson's disease, drug effect, or structural lesion must be excluded), imaging adds discriminatory value that clinical assessment alone cannot provide. In this scenario, the specific question is whether there is presynaptic dopaminergic terminal degeneration — answered by DAT-SPECT.
Scenario 2 — Suspected secondary or structural cause: When there are features suggesting a symptomatic (secondary) parkinsonism — hemiparkinsonism with a history of head trauma, vascular risk factors and stepwise progression, a brain mass on clinical assessment — structural imaging (MRI brain) is mandatory to exclude a mass lesion, subdural haematoma, hydrocephalus, or basal ganglia infarcts.
Scenario 3 — Drug-induced vs idiopathic PD distinction: When a patient on a dopamine-receptor blocking agent (DRBA) develops parkinsonism, the clinical question is: has the drug caused drug-induced parkinsonism (functional striatal dopaminergic deficit, reversible), or has the drug merely unmasked pre-existing idiopathic PD (structural nigrostriatal degeneration, irreversible)? This distinction has major therapeutic implications — stopping the drug resolves DIP but not PD. Nuclear medicine imaging (DAT-SPECT) can answer this question directly.
Scenario 4 — Parkinson-plus syndromes: When clinical features suggest MSA, PSP, or CBS rather than idiopathic PD, MRI brain can demonstrate specific structural changes (the hummingbird sign in PSP, the hot cross bun sign in MSA, cortical atrophy in CBS) that support the Parkinson-plus diagnosis and guide counselling about expected levodopa response and prognosis.
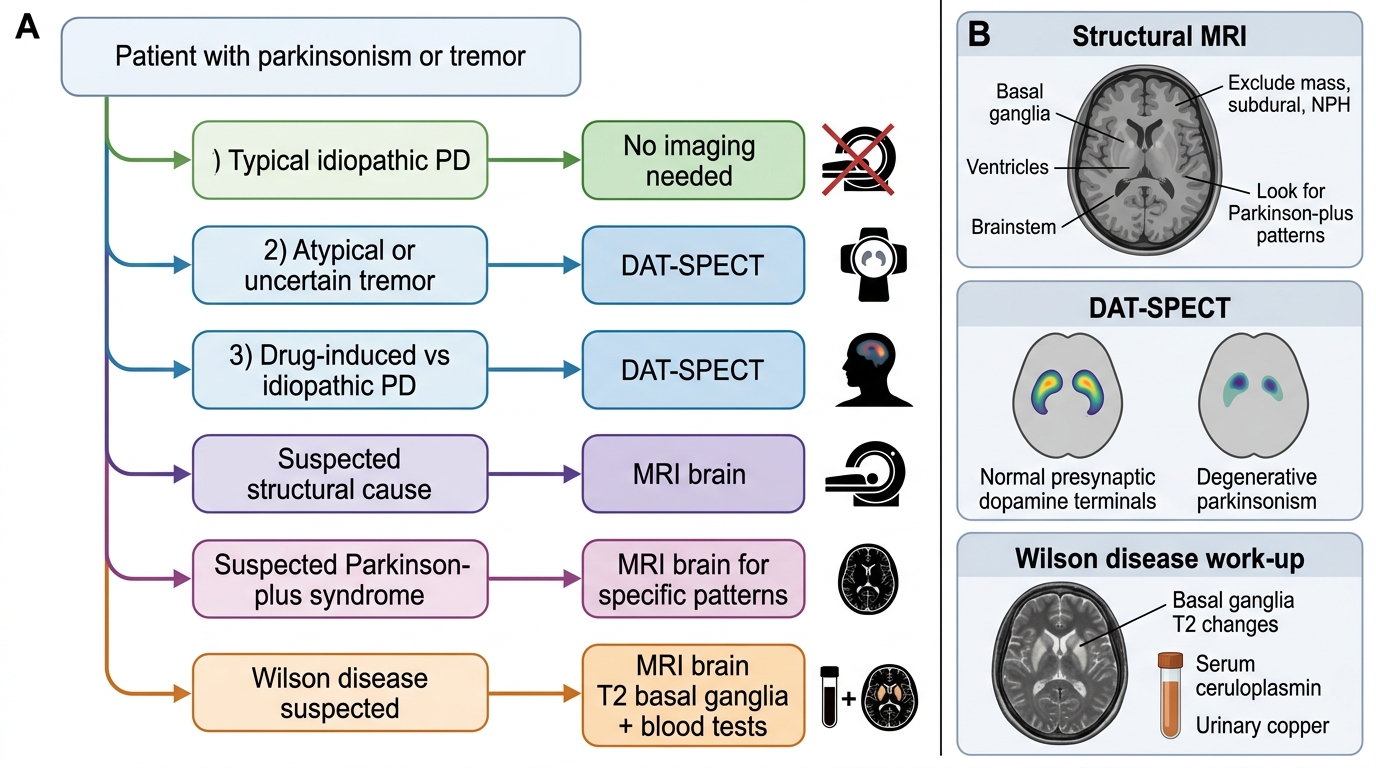
Imaging Decision Algorithm in Movement Disorders
Pathophysiology and Aetiology: Imaging Modalities and Their Interpretation
The imaging modalities used in movement disorders span structural (CT, MRI) and functional/nuclear medicine (DAT-SPECT, FDG-PET, MIBG scintigraphy) categories. Each modality measures a different biological substrate — structural integrity, presynaptic dopaminergic terminal density, cerebral metabolism, or cardiac sympathetic innervation — and has specific indications and interpretive requirements that must be understood in order to select and interpret them correctly.
Structural imaging:
CT brain has limited utility in idiopathic PD — it cannot visualise the substantia nigra or detect dopaminergic terminal loss. Its main use is to exclude structural lesions (space-occupying lesions, subdural haematoma, normal pressure hydrocephalus) in atypical presentations or when MRI is contraindicated. Calcification of the basal ganglia on CT may suggest Fahr disease (idiopathic basal ganglia calcification — a rare cause of movement disorder).
MRI brain provides superior soft-tissue contrast and is the imaging modality of choice for structural evaluation of movement disorders. Specific findings include:
- Parkinson's disease: standard MRI is typically normal in idiopathic PD. Substantia nigra changes (reduced neuromelanin signal on neuromelanin-sensitive MRI; reduced iron content on susceptibility-weighted imaging) are present but are research tools, not routinely used clinically.
- Progressive supranuclear palsy (PSP): midbrain atrophy on sagittal MRI produces the classic 'hummingbird sign' (or 'penguin sign') — a small midbrain with preserved pontine size, making the midbrain look like a hummingbird in profile; confirmed by the midbrain-to-pons ratio on MRI.
- Multiple system atrophy (MSA): cruciform signal hyperintensity in the pons on T2-weighted MRI, known as the 'hot cross bun sign' — reflecting pontine white matter degeneration with preservation of cortical spinal tracts; putaminal atrophy with a lateral putaminal T2 rim is also characteristic.
- Wilson's disease: T2 hyperintensity in the basal ganglia (putamen and caudate), thalamus, and midbrain; the classic 'face of the giant panda' sign refers to T2 hyperintensity in the midbrain tegmentum surrounding the normally dark red nucleus, resembling panda markings on axial T2 through the midbrain level.
- Vascular parkinsonism: multiple lacunar infarcts in the basal ganglia or white matter; may produce a distinctive 'lower body parkinsonism' pattern (prominent gait disorder, less upper limb involvement, minimal tremor).
- Normal pressure hydrocephalus (NPH): ventriculomegaly disproportionate to cortical atrophy on CT/MRI; presents with the triad of gait apraxia (magnetic gait), urinary incontinence, and cognitive decline (Hakim's triad) — mimics parkinsonism but responds to CSF drainage.
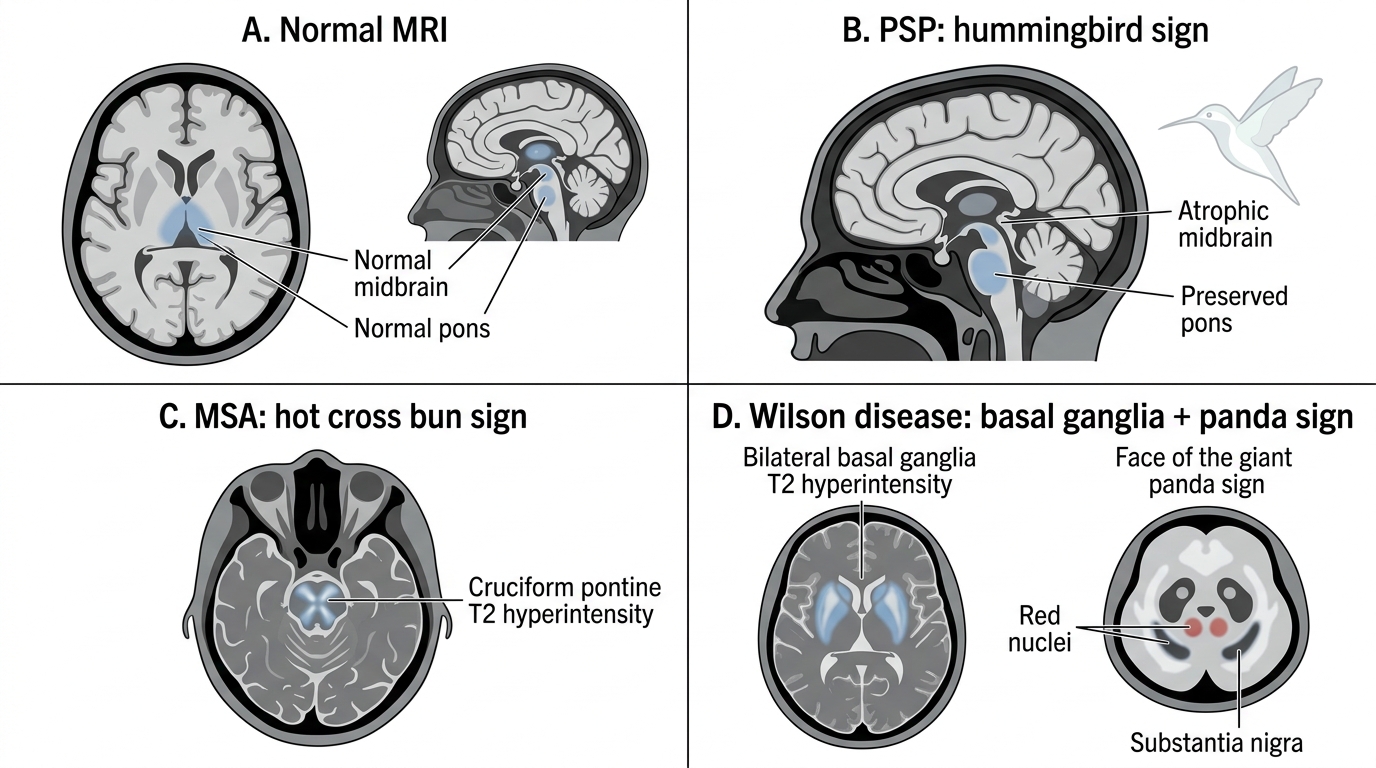
MRI Signs in Parkinsonian and Movement Disorders
Nuclear medicine imaging:
DAT-SPECT (DaTscan) uses a radiolabelled cocaine analogue (ioflupane-123I) that binds to the dopamine transporter (DAT) on presynaptic nigrostriatal terminals. In idiopathic PD, there is progressive loss of nigrostriatal terminals and reduced DAT binding, producing reduced radiotracer uptake in the striatum, particularly the putamen. The normal brain shows two symmetrical 'comma-shaped' areas of high uptake in the caudate and putamen; in PD, the posterior putamen shows reduced uptake first (asymmetric), then progresses to involve the whole putamen and later the caudate — producing a 'full stop' appearance.
Key interpretation principles:
- Idiopathic PD, PSP, MSA, CBS: ALL show reduced striatal DAT binding — because all involve presynaptic dopaminergic terminal loss. DAT-SPECT cannot distinguish between these conditions; it confirms presynaptic dopaminergic deficit.
- Drug-induced parkinsonism (DIP): NORMAL DAT-SPECT — DRBAs block postsynaptic D2 receptors but do NOT destroy presynaptic terminals; the terminals remain intact and bind the radiotracer normally. This is the critical clinical use: a normal scan in a patient with parkinsonism on a DRBA confirms DIP rather than pre-existing PD.
- Essential tremor: NORMAL DAT-SPECT — confirming no presynaptic dopaminergic deficit.
- DAT-SPECT is not affected by levodopa or dopamine agonist treatment — these act postsynaptically and do not alter DAT binding, making DAT-SPECT interpretable even in treated patients.
FDG-PET (fluorodeoxyglucose positron emission tomography): measures cerebral glucose metabolism. More useful in Parkinson-plus differentiation and DLB (marked temporoparietal hypometabolism in DLB, similar to Alzheimer's) than in routine PD diagnosis.
MIBG scintigraphy (meta-iodobenzylguanidine myocardial scintigraphy): MIBG is taken up by noradrenergic sympathetic nerve terminals on the myocardium. In PD and DLB, cardiac sympathetic denervation occurs early, producing reduced myocardial MIBG uptake. This distinguishes PD/DLB from MSA (cardiac sympathetic innervation is preserved in early MSA) — clinically useful when the DLB vs MSA distinction is needed.
SELF-CHECK
A 58-year-old man develops symmetric bilateral tremor and bradykinesia 6 months after starting haloperidol for delusional disorder. His psychiatrist wants to know whether this is drug-induced parkinsonism or idiopathic PD unmasked by haloperidol. Which investigation BEST discriminates these two diagnoses?
A. CT brain to look for basal ganglia lesions
B. MRI brain with susceptibility-weighted imaging
C. DAT-SPECT (DaTscan) to assess presynaptic dopaminergic terminal integrity
D. FDG-PET to assess cerebral glucose metabolism in the putamen
Reveal Answer
Answer: C. DAT-SPECT (DaTscan) to assess presynaptic dopaminergic terminal integrity
DAT-SPECT is the investigation of choice for distinguishing drug-induced parkinsonism (DIP) from idiopathic PD. In DIP, haloperidol blocks postsynaptic D2 receptors but does not destroy presynaptic dopaminergic terminals — so DAT binding (which measures presynaptic terminal integrity) is NORMAL. In idiopathic PD, there is progressive nigrostriatal terminal degeneration → reduced DAT binding → abnormal (reduced) DAT-SPECT. CT brain cannot visualise dopaminergic pathways. MRI-SWI shows nigrosome changes in PD but is not validated for DIP vs PD distinction in routine clinical use. FDG-PET shows metabolic changes in PD but does not reliably distinguish DIP from PD.
Diagnosis and Investigation: Integrating Blood Tests and Imaging
The practical interpretation of investigations in movement disorders requires integrating imaging findings with the clinical context. No investigation should be interpreted in isolation from the clinical picture — a normal DAT-SPECT in the context of clear clinical parkinsonism is more likely to reflect drug-induced parkinsonism than to be a false negative, while an abnormal DAT-SPECT must be contextualised against the clinical features to determine the specific cause. This section consolidates the full investigative approach to common movement disorder presentations, covering the blood tests and electrophysiology that complement imaging. This principle — that the investigation is a tool for answering a specific clinical question, not a checklist to tick — is what separates efficient clinical decision-making from unfocused over-investigation.
Blood tests in movement disorders:
Blood investigations are not part of the routine work-up for typical idiopathic PD but are critical for excluding secondary and reversible causes:
- Wilson's disease panel: serum ceruloplasmin (low in Wilson's — normal is 20–40 mg/dL; reduced to <20 mg/dL in Wilson's, though note it is an acute-phase reactant and may be falsely normal in acute liver disease); 24-hour urinary copper (elevated — >100 μg/24h in symptomatic patients, >250 μg/24h is considered diagnostic); liver function tests (transaminitis, coagulopathy in hepatic Wilson's). Confirm with ATP7B gene sequencing or liver biopsy copper quantification (>250 μg/g dry weight is the gold standard diagnostic threshold).
- Thyroid function: hyperthyroidism causes enhanced physiological tremor and occasionally frank movement disorder; hypothyroidism is associated with bradykinesia, slowed mentation, and cerebellar ataxia. TSH is an inexpensive screening test.
- Anti-streptococcal antibodies (ASOT, anti-DNase B): elevated in Sydenham's chorea from recent group A streptococcal infection; note that ASOT may normalise rapidly after penicillin treatment, so a normal ASOT does not exclude recent infection.
- Anti-phospholipid antibodies (APA, lupus anticoagulant): elevated in chorea associated with SLE and anti-phospholipid antibody syndrome (APAS); relevant in young women with chorea.
- Paraneoplastic antibody panel (anti-NMDAR, anti-Hu, anti-Yo, anti-CV2, anti-Ma2): indicated when a movement disorder develops subacutely in the setting of a suspected neoplasm (lung, breast, ovarian, testicular) or following a viral illness.
- Complete blood count with peripheral smear: acanthocytes (spiculated red cells) in neuroacanthocytosis — a rare genetic cause of chorea with orofacial self-mutilation; elevated CK in some myopathic movement disorder mimics.
Genetic testing:
Indicated when family history suggests a hereditary movement disorder, or when onset is unusually young (<50 for parkinsonism). LRRK2 (most common autosomal dominant PD mutation, 1–2% sporadic cases); PINK1/Parkin (autosomal recessive early-onset PD); GBA mutation (glucocerebrosidase, risk factor for PD and DLB); HTT CAG repeat count (Huntington's — >36 repeats disease-causing, >40 fully penetrant); ATP7B (Wilson's); DYT1/TOR1A (primary generalised dystonia in children/young adults).
Cerebrospinal fluid (CSF):
Routinely normal in idiopathic PD and essential tremor. Indicated when an inflammatory or infectious cause is suspected (encephalitis — herpes, autoimmune). CSF 14-3-3 protein and RT-QuIC (real-time quaking-induced conversion assay) are used in suspected prion disease (CJD — very rapid dementia + myoclonus + parkinsonism + periodic EEG discharges).
EEG and EMG:
EEG in myoclonus: cortical myoclonus shows high-amplitude cortical spike correlates on EEG-EMG coherence; specific patterns in prion disease and epileptic myoclonus. Surface EMG can measure tremor frequency and characterise the activation pattern (alternating agonist-antagonist vs co-contraction).
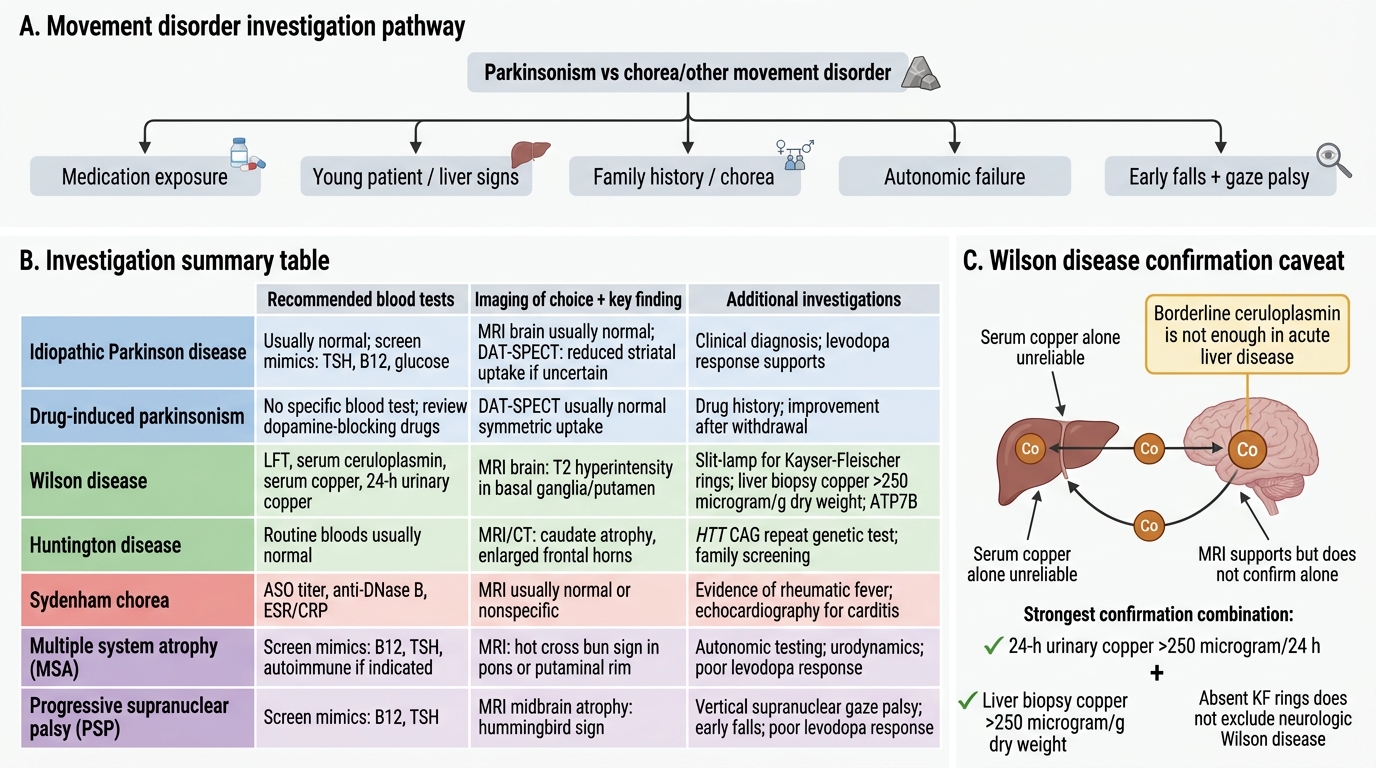
Investigations for Movement Disorders
SELF-CHECK
In a patient with suspected neurological Wilson's disease, serum ceruloplasmin is 18 mg/dL (reference 20–40 mg/dL). The patient also has jaundice and transaminitis suggesting acute hepatic disease. Which combination of findings would MOST reliably confirm the diagnosis?
A. Serum copper alone — Wilson's disease always shows low serum copper
B. 24-hour urinary copper >250 μg/24h AND liver biopsy copper >250 μg/g dry weight
C. MRI brain showing T2 hyperintensity in the putamen — sufficient for diagnosis
D. Absence of Kayser-Fleischer rings excludes neurological Wilson's disease
Reveal Answer
Answer: B. 24-hour urinary copper >250 μg/24h AND liver biopsy copper >250 μg/g dry weight
In acute liver disease, ceruloplasmin may be borderline or normal (it is an acute-phase reactant), so a single borderline ceruloplasmin is not sufficient for confirmation. The combination of elevated 24-hour urinary copper (>250 μg/24h is considered diagnostic in symptomatic patients) AND liver biopsy copper quantification (>250 μg/g dry weight = gold standard) provides the most reliable biochemical confirmation. Serum copper alone is unreliable (can be normal or elevated, reflecting free/non-ceruloplasmin-bound copper). MRI findings (basal ganglia T2 changes, face of the giant panda) are supportive but not diagnostic. Note: Absence of Kayser-Fleischer rings does NOT exclude Wilson's disease — KF rings are virtually universal in neurological Wilson's but may be absent in isolated hepatic presentation without neurological involvement.