Page 13 of 27
IM22.7 | Phosphide Poisoning — SDL Guide
Learning Objectives
- Describe the toxicology and mechanism of toxicity of aluminium phosphide and zinc phosphide
- Enumerate the clinical features of phosphide poisoning and their pathophysiological basis
- Explain why there is NO specific antidote for aluminium phosphide poisoning and describe the principles of supportive management
- Describe the prognosis of aluminium phosphide poisoning and identify the markers of severity
- Outline the role of coconut oil and other investigational measures in phosphide poisoning
INSTRUCTIONS
Aluminium phosphide (AlP, celphos) poisoning carries one of the highest case fatality rates of any acute poisoning in India, approaching 70-100% in severe cases. This module covers the toxicology, clinical course, lack of specific antidote, and the critical importance of supportive care and early recognition of severity markers.
References
- Harrison's Principles of Internal Medicine, 21st ed., Ch. 454 — Poisoning and Drug Overdose (textbook)
- API Textbook of Medicine, 10th ed., Ch. 26 — Poisoning (textbook)
- Davidson's Principles and Practice of Medicine, 24th ed., Ch. 9 — Poisoning (textbook)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
It is 3 am in a district hospital in Punjab. A 25-year-old man is brought in by his family after consuming two tablets of a silver-grey pesticide used for grain storage. The family found the tablet wrapper — Celphos 56% tablets, aluminium phosphide. He is vomiting continuously and complaining of severe epigastric pain. His blood pressure is 90/60 mmHg and pulse is 115 bpm. You know there is no antidote. You know that if he absorbed a significant dose, the mortality approaches 70–100% even with the best supportive care. You also know that the next decisions — what NOT to do, what to monitor, and how to prepare for the cascade of cardiac, respiratory, and metabolic failure that is about to unfold — are entirely within your control. There are no specific antidotes on the hospital shelf. There is no miracle drug. There is only the quality of your resuscitative thinking, your knowledge of the pathophysiology, and your ability to support failing organ systems while the phosphine gas that is already in his bloodstream destroys mitochondria across every organ. This module prepares you for that night.
WHY THIS MATTERS
Aluminium phosphide (AlP) poisoning is the single most lethal acute poisoning encountered in Indian emergency departments, accounting for a disproportionate share of poisoning deaths in agricultural northern and central India (Punjab, Haryana, Rajasthan, Madhya Pradesh). Celphos tablets (aluminium phosphide 56%) are widely used as a grain fumigant and rodenticide and are readily available in agricultural communities. The deliberate ingestion of AlP as a means of suicide is distressingly common and carries a case fatality rate of 60–80% or higher, even in tertiary care hospitals, because there is no specific antidote and the toxin causes direct mitochondrial toxicity that is not amenable to pharmacological reversal. Zinc phosphide is used as a rodenticide and has a similar mechanism but is generally less toxic due to lower potency. As a physician in any Indian rural or urban posting, you will encounter phosphide poisoning and must be prepared to manage it with the tools available: rigorous supportive care, avoidance of harmful interventions, and clear prognostic communication to the family.
RECALL
Recall from biochemistry the role of mitochondria in cellular energy production: the electron transport chain (ETC) complexes I–IV use electrons from NADH and FADH₂ to pump protons across the inner mitochondrial membrane, creating a proton gradient that drives ATP synthase (Complex V). Any agent that disrupts this proton gradient — by either blocking the ETC or creating an alternative proton leak across the membrane — prevents ATP synthesis from the proton gradient and causes cellular energy failure. This is called uncoupling of oxidative phosphorylation when the proton gradient is dissipated without driving ATP synthase. From physiology, recall that the myocardium is entirely dependent on aerobic metabolism (cardiac muscle has minimal anaerobic capacity) — this explains why the heart is the primary target organ in phosphide poisoning. Recall from pharmacology that there is no drug that can restore the mitochondrial function of cells already poisoned by phosphine.
Clinical Presentation of Aluminium Phosphide Poisoning
Aluminium phosphide poisoning produces a characteristic clinical course that unfolds in a predictable sequence over hours, progressing from initial gastrointestinal features through cardiovascular collapse to multi-organ failure. Recognising the early features allows the physician to anticipate the subsequent phases, prepare for cardiovascular resuscitation, and counsel the family regarding prognosis before the patient deteriorates beyond reversibility.
The first clinical features appear within 30 minutes to 4 hours of ingestion, as AlP reacts with gastric acid and atmospheric moisture to liberate phosphine gas (PH₃). The onset is faster with greater gastric acidity, larger tablet dose, and co-ingestion of liquids (which accelerate tablet dissolution). Early features are primarily gastrointestinal: severe nausea, profuse vomiting (often described as having a garlic-like or fishy odour from the phosphine), epigastric and abdominal pain, and diarrhoea. These symptoms reflect both direct mucosal injury from phosphine and the systemic absorption that is already occurring.
Cardiovascular features are the dominant and life-threatening manifestation, typically developing within 4–12 hours of ingestion. The sequence is: tachycardia (early, compensatory — from hypotension and sympathetic activation) → hypotension (from myocardial dysfunction, vasodilation, and distributive shock from phosphine-mediated endothelial injury) → refractory shock (cardiogenic + distributive components, not responding to fluids or vasopressors) → multi-organ failure and death. Cardiac arrhythmias are prominent and varied: sinus tachycardia initially; then ST-segment changes and T-wave abnormalities; progressing to atrial fibrillation, ventricular tachycardia, and VF in the terminal phase. The ECG changes reflect direct myocardial mitochondrial toxicity — every cardiomyocyte is energy-depleted.
Respiratory features: Acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) develop from direct phosphine toxicity to pulmonary endothelium, causing non-cardiogenic pulmonary oedema. Tachypnoea, cyanosis, and oxygen-refractory hypoxia are late and catastrophic signs.
Metabolic features: Severe metabolic acidosis (lactic acidosis from cellular energy failure across all tissues), hyperglycaemia or hypoglycaemia, and electrolyte disturbances. Serum lactate is a useful severity marker — persistently rising lactate despite resuscitation indicates irreversible cellular injury and very high mortality.
Neurological features: Headache, dizziness, and agitation are early; as hypotension worsens, drowsiness, delirium, seizures, and coma develop. These are predominantly consequences of cerebral hypoperfusion rather than direct CNS phosphine toxicity.

Clinical Timeline of Aluminium Phosphide Poisoning
Toxicology and Pathophysiology of Phosphine Toxicity
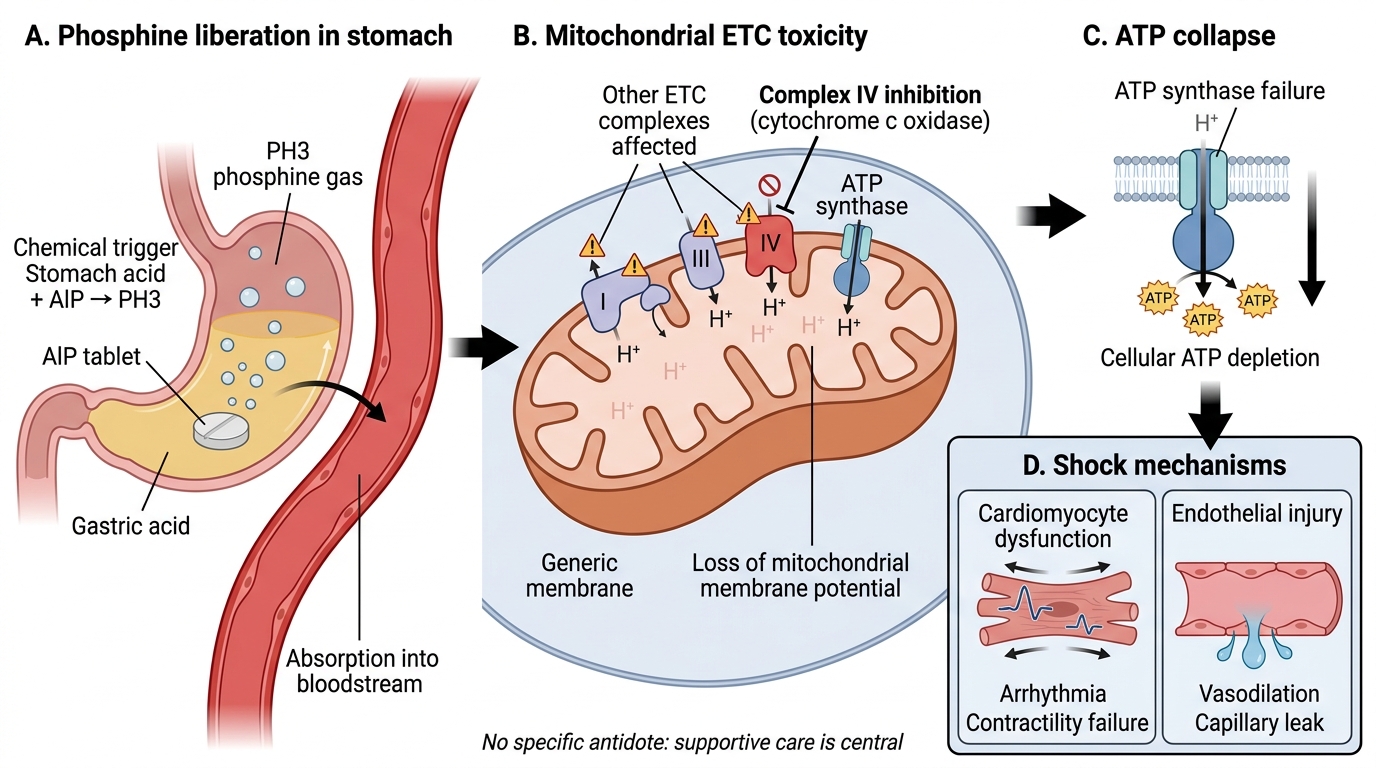
The toxicology of aluminium phosphide poisoning is driven by the spontaneous liberation of phosphine gas (PH₃) when AlP reacts with moisture, and by the potent mitochondrial toxicity of phosphine once absorbed. Understanding this mechanism explains both the clinical syndrome and — crucially — why there is no specific antidote and why most supportive interventions are directed at consequences rather than the cause.
The chemical reaction: Aluminium phosphide reacts with water (from gastric acid, atmospheric humidity, or moisture in grain stores) according to: AlP + 3H₂O → Al(OH)₃ + PH₃. Phosphine gas is also spontaneously liberated at room temperature in the presence of moisture. This means: (1) the tablet continues to generate phosphine in the stomach as long as it is intact; (2) belching or vomiting releases phosphine-laden gas — creating a risk of secondary inhalation for bystanders and healthcare workers (respirators should be worn in the resuscitation room); (3) gastric lavage is controversial — it may accelerate the liberation of phosphine by increasing the contact surface area between tablet and gastric fluid, but may also remove intact tablet, reducing total absorbed dose. Vomiting and gastric lavage increase phosphine exposure for healthcare workers in the room.
Mechanism of cellular toxicity: Phosphine is a potent inhibitor of cytochrome c oxidase (Complex IV of the electron transport chain) — the same enzyme inhibited by cyanide, but phosphine also inhibits multiple other respiratory chain complexes (I, II, and IV) and directly damages mitochondrial membranes via lipid peroxidation. The result is: blocked electron transport → no proton pumping → loss of mitochondrial membrane potential → failure of ATP synthase → cellular ATP depletion. In addition, phosphine generates reactive oxygen species (ROS) that cause oxidative damage to proteins, lipids, and DNA. This combination of ETC inhibition and oxidative stress is why the damage is so rapid and so irreversible — cells cannot repair themselves without the ATP they cannot produce.
Why the heart is the primary target: Cardiac myocytes have the highest mitochondrial density of any cell in the body and are entirely aerobic — they have no glycolytic reserve to compensate for mitochondrial failure. Phosphine-mediated ATP depletion in cardiomyocytes rapidly causes: impaired contractility (reduced cardiac output), impaired relaxation (diastolic dysfunction), disrupted membrane ion channel function (arrhythmias from abnormal electrical conduction), and eventually cardiomyocyte death. The resulting pattern — cardiogenic shock with myocarditis-like features, combined with distributive shock from systemic endothelial injury — is uniquely resistant to standard vasopressor therapy because the underlying problem is cellular energy failure, not vascular tone or volume depletion.
Zinc phosphide mechanism: Zinc phosphide (Zn₃P₂) reacts with gastric acid to release phosphine: Zn₃P₂ + 6HCl → 3ZnCl₂ + 2PH₃. The mechanism is identical to AlP. However, zinc phosphide releases phosphine more slowly (lower potency per gram), typically at a lower effective dose per ingested quantity, making it somewhat less uniformly fatal. The clinical syndrome is similar but may be less severe for the same tablet count.
No specific antidote — mechanistic explanation: Unlike organophosphate poisoning (where the enzyme inhibition is partially reversible) or cyanide poisoning (where hydroxocobalamin directly binds cyanide and excretes it), phosphine causes direct oxidative damage to mitochondria that is not enzymatically reversible. There is no drug that can restore the phosphorylation capacity of damaged mitochondria, chelate intracellular phosphine, or replace the cells that have already undergone energy failure-mediated death. Supportive therapy aims to maintain organ perfusion (using vasopressors, intra-aortic balloon pump in refractory cardiogenic shock) and limit ongoing oxidative damage (antioxidant strategies, though not proven to reduce mortality).
Aluminium Phosphide Poisoning: Phosphine-Induced Mitochondrial Failure
SELF-CHECK
A 30-year-old farmer is brought to casualty 2 hours after ingesting 2 AlP tablets. He is vomiting, BP 80/50 mmHg, HR 130 bpm, SpO₂ 90%, serum lactate 8.2 mmol/L. His family asks if there is a specific antidote. Which explanation is MOST accurate?
A. Pralidoxime 2 g IV can partially reverse the mitochondrial toxicity of phosphine
B. N-acetylcysteine IV can replenish glutathione and neutralise phosphine-induced oxidative damage
C. There is no specific antidote; phosphine causes irreversible direct mitochondrial damage not amenable to pharmacological reversal
D. Desferrioxamine can chelate phosphine from the bloodstream and should be given immediately
Reveal Answer
Answer: C. There is no specific antidote; phosphine causes irreversible direct mitochondrial damage not amenable to pharmacological reversal
Aluminium phosphide poisoning has NO specific antidote. Phosphine inhibits multiple mitochondrial electron transport chain complexes (principally Complex IV, cytochrome c oxidase) and causes direct oxidative damage to mitochondrial membranes via lipid peroxidation. This cellular injury is not enzymatically reversible and cannot be pharmacologically chelated or neutralised. Pralidoxime reactivates inhibited acetylcholinesterase (for OP poisoning — entirely different mechanism). NAC replenishes glutathione (for paracetamol/NAPQI toxicity). Desferrioxamine chelates iron (not phosphine). The correct management is aggressive supportive care targeting haemodynamic stabilisation, oxygenation, and monitoring for multi-organ failure.
Diagnosis and Severity Assessment
Diagnosis of aluminium phosphide poisoning is predominantly clinical and historical. The combination of a history of ingesting a grain fumigant or rodenticide tablet (silver-grey, white, or yellow tablet found in the agricultural setting), together with the acute onset of vomiting with garlic-like odour and early cardiovascular instability, is virtually diagnostic in the Indian context. Investigations serve to confirm systemic absorption, quantify organ damage, and provide severity markers that guide prognosis and resource allocation — including the decision of when to initiate intensive care measures and when to counsel the family about the limits of what medicine can achieve.
There is no bedside test that directly measures phosphine concentration in blood — diagnosis relies on clinical and circumstantial evidence. The silver nitrate paper test (a bedside test where a strip of 10% silver nitrate paper placed over the patient's vomitus turns black-brown in the presence of phosphine gas) is a practical diagnostic aid used in some centres. It is not uniformly available but is highly specific when positive.
Key investigations:
- ABG (urgent): Metabolic acidosis (high anion gap lactic acidosis) is present from the earliest stages. Severity: pH < 7.2 indicates severe poisoning; pH < 7.0 is associated with near-universal mortality.
- Serum lactate: Rising lactate is the most important real-time severity marker. Persistent or rising lactate despite fluid resuscitation indicates cellular energy failure and very high mortality. Serial lactate measurements every 2–4 hours.
- ECG (continuous monitoring): Sinus tachycardia (early) → ST depression or elevation (myocardial ischaemia/injury) → QTc prolongation → atrial or ventricular arrhythmias (VT, VF). The ECG deterioration pattern correlates with severity and predicts cardiac arrest.
- Echocardiography (where available): Global hypokinesis and reduced ejection fraction confirm cardiomyopathy from phosphine toxicity; biventricular failure is common in severe poisoning.
- Serum electrolytes: Hypokalaemia, hypomagnesaemia (arrhythmia risk); hyperglycaemia from stress response; or hypoglycaemia in severe liver involvement.
- Renal function and liver function: AKI from renal tubular hypoxia; elevated transaminases from hepatic injury. Both are late complications and markers of multi-organ involvement.
- Haemogram: Leucocytosis from stress; thrombocytopaenia in DIC (disseminated intravascular coagulation) in terminal phase.
- Chest X-ray: Bilateral infiltrates of ARDS; pulmonary oedema.
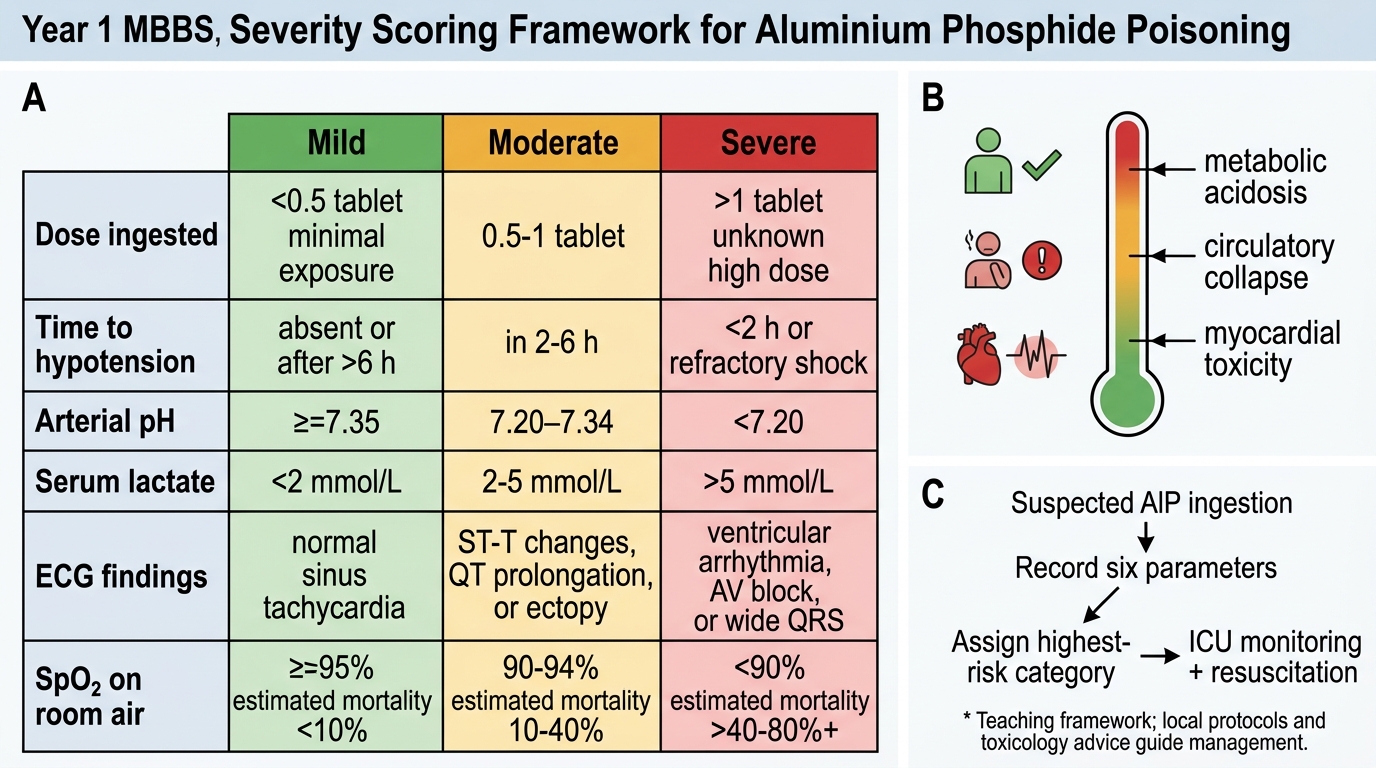
Prognostic markers (predict high mortality):
- Dose: ingestion of more than 1 AlP tablet (500 mg or more of AlP) is associated with high mortality; 2 or more tablets = very high mortality
- Early cardiovascular collapse (hypotension within 4 hours)
- Persistent metabolic acidosis (pH < 7.2)
- Serum lactate > 8 mmol/L and not responding to resuscitation
- QRS widening, VT, or complete heart block on ECG
- Rapidly worsening SpO₂ requiring high FiO₂
Severity Scoring in Aluminium Phosphide Poisoning