Page 10 of 17
IM23.4-7 | Sodium and Potassium Disorders — SDL Guide
Learning Objectives
- Enumerate the causes of hyponatraemia and hypernatraemia and describe the correct diagnostic and management approach for each
- Enumerate the causes and describe the clinical and laboratory features of hypokalaemia and hyperkalaemia
- Outline the management of hyperkalaemia including the sequence and rationale of each intervention
- Identify the risks of overcorrection in hyponatraemia (osmotic demyelination) and hyperkalaemia cardiac emergencies
INSTRUCTIONS
Sodium and potassium are the dominant extracellular and intracellular cations respectively. Disorders of these two electrolytes are among the most common and consequential problems in hospital medicine, contributing directly to altered consciousness, cardiac arrhythmias, respiratory failure, and death. This module takes the IM-foundation arc: establishing normal physiology first, then building mechanistically to the clinical disorders and their management.
References
- Harrison's Principles of Internal Medicine, 21st ed., Ch. 49 — Fluid and Electrolyte Disturbances (textbook)
- API Textbook of Medicine, 10th ed. — Electrolyte Disorders (textbook)
- Davidson's Principles and Practice of Medicine, 24th ed., Ch. 19 — Water and Electrolyte Disturbances (textbook)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
Ward round, 7 AM. You are handed two cards:
Patient A: 72-year-old woman admitted for community-acquired pneumonia. She is on intravenous antibiotics and has been 'a bit confused' overnight. Sodium: 118 mmol/L. Yesterday it was 136 mmol/L. The night team started her on hypertonic saline. By 6 AM she had received 250 mL of 3% saline. Her sodium is now 134 mmol/L — a correction of 16 mmol/L in 18 hours. You realise with alarm that this almost certainly exceeds the safe limit.
Patient B: 55-year-old man with chronic kidney disease (eGFR 24) admitted for a foot cellulitis. Potassium from the evening panel: 6.8 mmol/L. His ECG shows peaked T waves and a PR interval of 240 ms. His registrar has written 'monitor repeat K tomorrow morning'.
Both patients are in immediate danger from electrolyte emergencies that are not being treated correctly. Patient A risks osmotic demyelination syndrome from overcorrection. Patient B risks ventricular fibrillation within hours. Understanding the physiology, limits, and emergency management of sodium and potassium disorders is not optional for a physician managing ward patients.
WHY THIS MATTERS
Electrolyte disorders are the bread-and-butter of every medical ward. Hyponatraemia is the most common electrolyte abnormality in hospitalised patients, occurring in 15–30% of admissions, and is independently associated with increased in-hospital mortality. Hyperkalaemia is the most immediately life-threatening electrolyte disorder — it can kill within minutes by causing ventricular fibrillation in a patient who was haemodynamically stable an hour earlier. For NMC competencies IM23.4–23.7, you must be able to enumerate causes, describe clinical features, and work through the diagnostic algorithm correctly — but most importantly, you must recognise when an electrolyte disorder constitutes a medical emergency requiring immediate treatment, and you must know the safe limits and the correct sequencing of interventions.
RECALL
Activate your knowledge of body fluid compartments and renal physiology. Total body water (TBW) in a 70 kg adult is approximately 42 litres (60% of body weight in men, 50–55% in women). Of this, two-thirds (28 L) is intracellular fluid (ICF) and one-third (14 L) is extracellular fluid (ECF). ECF is divided into plasma (3.5 L) and interstitial fluid (10.5 L). The major cation of the ECF is sodium (normal serum 135–145 mmol/L); the major cation of the ICF is potassium (intracellular K⁺ ≈ 140 mmol/L; serum K⁺ normal 3.5–5.0 mmol/L). Serum sodium is the primary determinant of plasma osmolality, calculated as: serum osmolality = 2 × Na + glucose/18 + urea/2.8 (in SI units: 2 × Na + glucose + urea). Normal serum osmolality is 275–295 mOsm/kg. Antidiuretic hormone (ADH), also known as arginine vasopressin, is released from the posterior pituitary in response to rising osmolality (detected by hypothalamic osmoreceptors) or falling blood volume/pressure (detected by carotid baroreceptors). ADH acts on V2 receptors in the renal collecting duct to insert aquaporin-2 water channels, increasing water reabsorption. The sodium concentration is therefore ultimately a ratio of total body sodium to total body water, not simply a measure of the sodium content alone. Potassium is regulated primarily by the renin-angiotensin-aldosterone system (RAAS): aldosterone increases potassium secretion in the principal cells of the collecting duct via the Na⁺/K⁺-ATPase and ROMK channels.
Orientation: Sodium as a Marker of Water Balance
The clinical approach to sodium disorders requires a conceptual shift: serum sodium reflects the balance between total body sodium and total body water, not the absolute sodium content. Hyponatraemia (Na <135 mmol/L) means there is too much water relative to sodium — not necessarily that sodium is depleted. Hypernatraemia (Na >145 mmol/L) means there is too little water relative to sodium — not necessarily that the body is sodium-overloaded. This reframing has direct practical implications: in most hyponatraemia, treatment involves restricting water or removing it, not simply infusing more sodium. In most hypernatraemia, treatment involves replacing free water, not giving sodium.
Potassium disorders are clinically significant because potassium is the principal determinant of the resting membrane potential across excitable cells (neurons, cardiac myocytes, skeletal muscle). The resting potential is governed by the ratio of intracellular to extracellular K⁺ concentration; a low serum potassium (hypokalaemia) hyperpolarises the membrane, making cells less excitable; a high serum potassium (hyperkalaemia) depolarises the membrane partially, initially increasing excitability but then inactivating sodium channels and producing inexcitable, flaccid cells. Both extremes cause dangerous cardiac and neuromuscular dysfunction, and both require careful management.
A critical technical point: the serum potassium reflects only 2% of total body potassium (the remainder is intracellular). A serum K⁺ of 3.0 mmol/L may therefore represent a total body deficit of 200–400 mmol. Conversely, shifts of potassium between compartments (e.g., from acidosis, insulin deficiency, or cell lysis) change serum K⁺ dramatically without any change in total body K⁺. Recognising transcellular shifts as a mechanism allows you to treat the right target — correcting the shift versus replacing the genuine deficit.
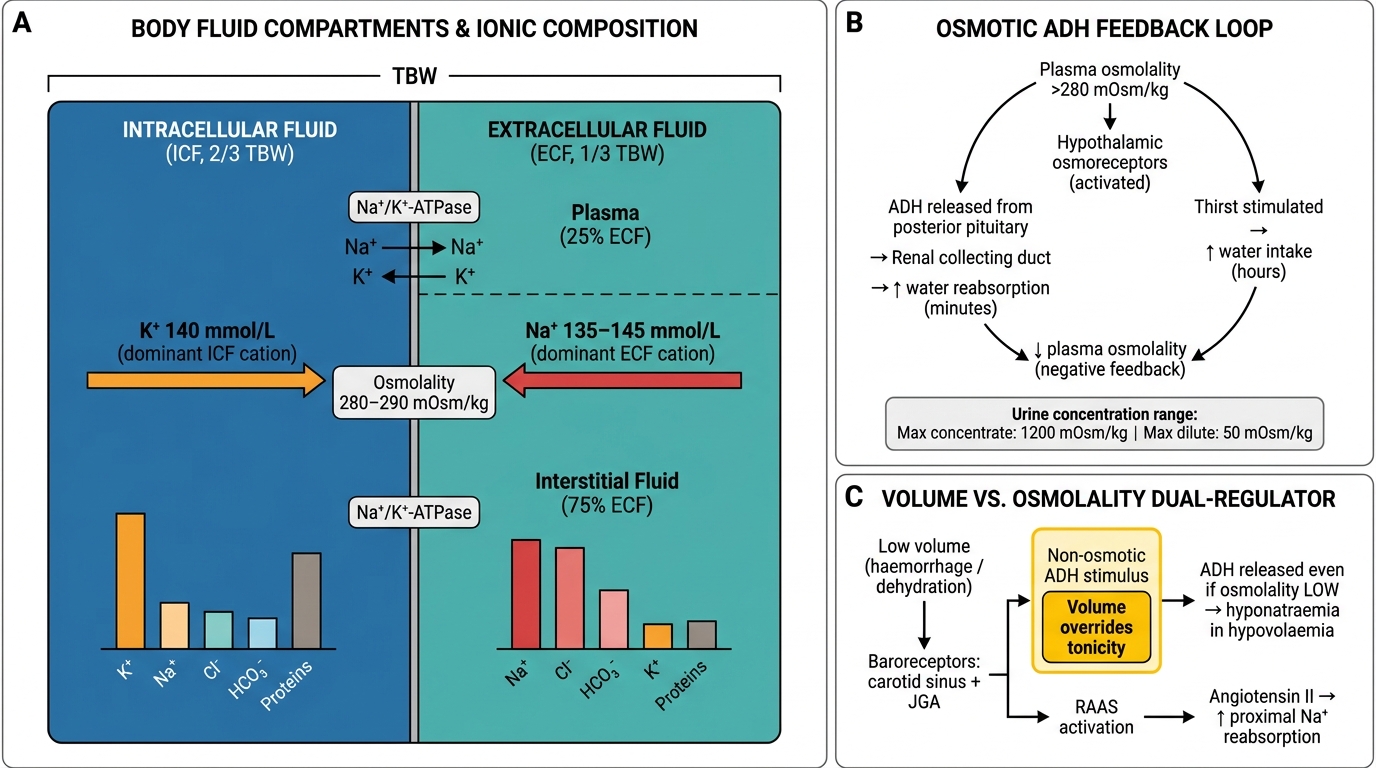
Body Fluid Compartments, Electrolyte Distribution, and ADH Regulation of Water Balance
Normal Sodium and Potassium Homeostasis
Normal serum sodium (135–145 mmol/L) is maintained by the interplay between water intake, ADH-driven water reabsorption, and thirst. The hypothalamic osmoreceptors detect a rise in plasma osmolality above approximately 280 mOsm/kg and trigger two responses simultaneously: release of ADH (increasing renal water reabsorption) and stimulation of thirst (increasing water intake). ADH acts within minutes; the thirst response operates over hours to days. The kidney can concentrate urine to a maximum of approximately 1200 mOsm/kg (requiring maximal ADH activity) and dilute it to as low as 50 mOsm/kg (in complete ADH suppression). This wide range of concentrating ability gives the kidney extraordinary flexibility in managing water excess or deficit.
Volume versus osmolality: the two independent regulators. Volume is sensed by baroreceptors in the carotid sinus and juxtaglomerular apparatus. When volume is low (haemorrhage, dehydration), baroreceptor-mediated ADH release is triggered even if osmolality is normal or low — this is the 'non-osmotic' ADH stimulus that explains why hyponatraemia develops in hypovolaemia (the body prioritises volume over tonicity). The RAAS also activates in volume depletion: angiotensin II increases proximal tubular sodium reabsorption and stimulates aldosterone, which increases collecting duct sodium reabsorption.
Potassium homeostasis involves two phases: (1) acute transcellular buffering — within minutes, approximately 98% of a potassium load is taken up into cells, buffered by insulin (activates Na⁺/K⁺-ATPase), catecholamines (via β₂ adrenergic receptors), and alkalosis (H⁺ moves out of cells in exchange for K⁺ moving in); (2) renal excretion — over hours, aldosterone drives K⁺ secretion in the principal cells of the cortical collecting duct via ROMK channels and Na⁺/K⁺-ATPase. The rate of urinary K⁺ excretion can be assessed by the transtubular potassium gradient (TTKG) or the spot urine potassium-to-creatinine ratio; a high ratio (>20 mmol/mmol or urine K⁺ >20 mmol/L) in a hypokalaemic patient indicates renal K⁺ wasting; a low ratio (<20 mmol/mmol or urine K⁺ <20 mmol/L) indicates extrarenal losses or poor intake.
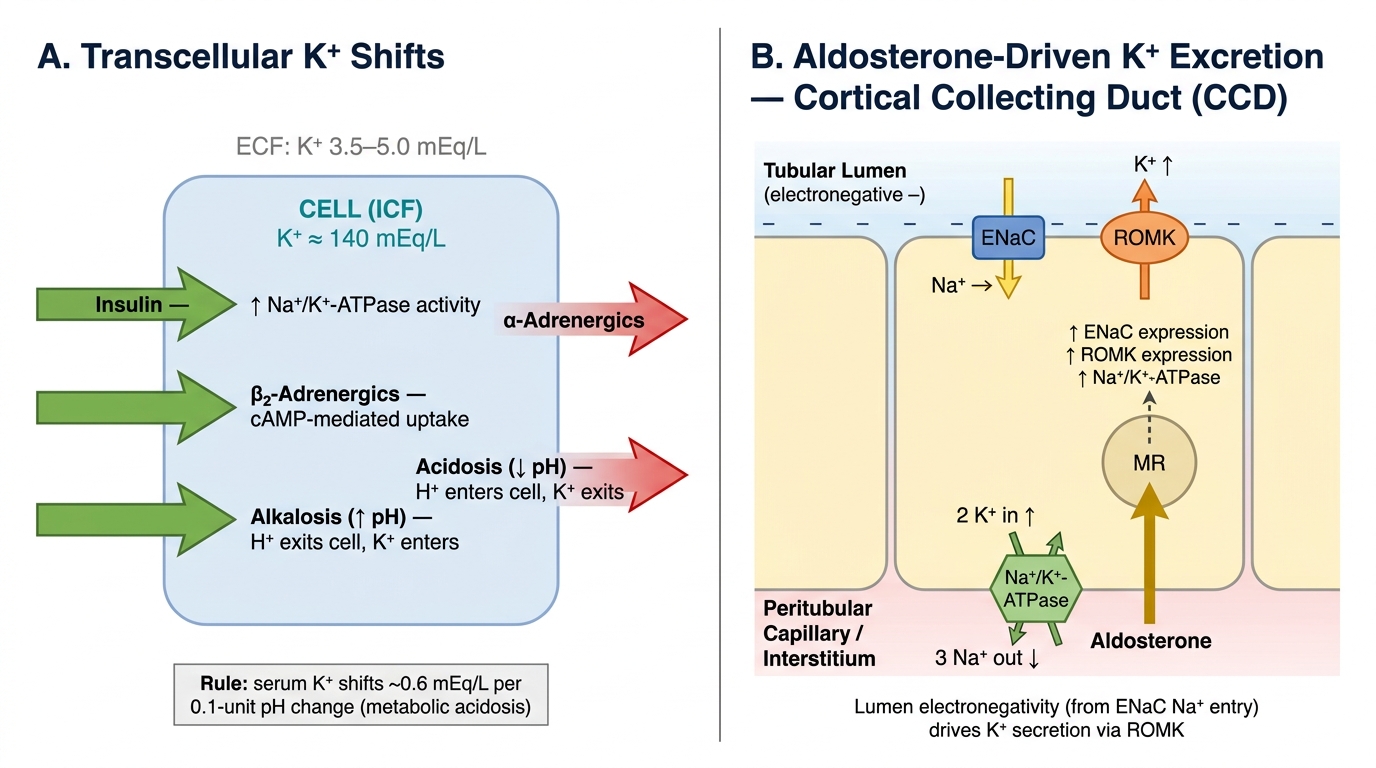
Potassium Homeostasis: Transcellular Shifts and Renal Excretion in the Cortical Collecting Duct
Hyponatraemia: Causes, Diagnosis, and Management
Hyponatraemia is defined as serum sodium <135 mmol/L. It is the most common electrolyte disorder in hospitalised patients, affecting 15–30% of admissions and carrying independent mortality risk. The correct diagnostic approach uses three sequential assessments: serum osmolality, volume status, and urine sodium plus urine osmolality. Understanding why each step is necessary is more important than memorising the algorithm — the physiology drives the logical sequence. The first step determines whether the hyponatraemia is genuine (hypotonic) or artifactual (isotonic — pseudohyponatraemia) or due to an osmotically active solute other than sodium (hypertonic — e.g., hyperglycaemia). Only hypotonic hyponatraemia requires further evaluation of the fluid compartment and urine chemistry. The second step identifies whether the body is sodium-depleted (hypovolaemic), sodium-normal (euvolaemic), or sodium-excess with proportionally greater water excess (hypervolaemic). The third step — urine sodium and osmolality — identifies the renal response, which localises the pathological ADH driver and guides fluid prescription. Many clinical errors in hyponatraemia management arise from skipping step 1 (giving saline to a patient with SIADH worsens hyponatraemia because the kidney excretes the sodium and retains the water) or from misclassifying volume status.
The three osmolality categories and their meaning:
- Isotonic/pseudohyponatraemia (serum osmolality normal, 275–295 mOsm/kg): occurs when space is displaced by protein (severe hyperproteinaemia, e.g., multiple myeloma) or fat (severe hypertriglyceridaemia), falsely lowering the measured sodium by older methods. Ionised calcium and glucose are normal; modern direct ISE analysers are not affected. No treatment required for sodium.
- Hypertonic hyponatraemia (serum osmolality >295 mOsm/kg): occurs when another osmotically active solute (most commonly glucose, or mannitol) draws water out of cells, diluting plasma sodium. The correction factor for hyperglycaemia: measured Na increases by approximately 1.6 mmol/L for every 5.6 mmol/L (100 mg/dL) rise in glucose. Treatment is directed at the primary disorder (glycaemia control).
- Hypotonic hyponatraemia (serum osmolality <275 mOsm/kg): the clinically significant category. All further steps apply here.
Step 2 — Volume status assessment (clinical: skin turgor, JVP, blood pressure, oedema):
- Hypovolaemic hyponatraemia (volume depleted — collapsed JVP, tachycardia, reduced skin turgor): sodium losses exceed water losses. Renal causes (urine Na >20 mmol/L): diuretics (especially thiazides — thiazides block NaCl reabsorption in the cortical diluting segment, impairing urinary dilution without disrupting the medullary concentrating gradient; unlike loop diuretics which wreck the medullary gradient, thiazides impair free-water clearance while preserving concentrating ability, causing water retention relative to sodium), salt-losing nephropathy, adrenal insufficiency (mineralocorticoid deficiency). Extrarenal causes (urine Na <20 mmol/L): vomiting, diarrhoea, third-space losses (burns, pancreatitis); kidneys retain sodium avidly. Management: isotonic (0.9%) saline to restore volume.
- Euvolaemic hyponatraemia (no oedema, no volume depletion): the most common category. The cause is almost always excess ADH with normal sodium content — too much water. Principal causes: SIADH (syndrome of inappropriate antidiuretic hormone secretion — see below), hypothyroidism (thyroid hormone normally downregulates renal ADH sensitivity), glucocorticoid deficiency (cortisol tonically inhibits hypothalamic ADH release — adrenal insufficiency removes this inhibition), and psychogenic polydipsia (excessive water intake overwhelms renal diluting capacity; urine is maximally dilute — osmolality <100 mOsm/kg). Management: fluid restriction ± specific therapy for the cause.
- Hypervolaemic hyponatraemia (oedematous — peripheral oedema, ascites, raised JVP): total body sodium is increased but water is increased proportionally more. Causes: congestive heart failure (low cardiac output activates baroreceptors → non-osmotic ADH + RAAS → avid salt and water retention), cirrhosis (splanchnic vasodilation → effective hypovolaemia → same activation), nephrotic syndrome and advanced CKD. Urine Na is usually <20 mmol/L (kidneys retain sodium avidly). Management: treat the underlying disease; fluid and sodium restriction; diuretics.
SIADH — diagnostic criteria (Schwartz-Bartter criteria): (1) Serum Na <135 mmol/L; (2) Serum osmolality <275 mOsm/kg (hypotonic); (3) Urine osmolality inappropriately concentrated (>100 mOsm/kg, usually >300) despite hypotonicity — kidneys continue excreting concentrated urine when they should be diluting; (4) Urine Na >40 mmol/L (kidneys excrete the extra sodium they receive); (5) Clinically euvolaemic; (6) Normal thyroid and adrenal function (must exclude these). Causes of SIADH: CNS disorders (meningitis, encephalitis, SAH, stroke, head injury), pulmonary disorders (pneumonia, TB, ARDS, mechanical ventilation), drugs (SSRIs, TCAs, carbamazepine, cyclophosphamide, NSAIDs, chlorpropamide, vincristine), malignancy (especially small cell lung carcinoma — most common cause of ectopic ADH).
Hyponatraemia management — the critical rate limit:
The danger in treating hyponatraemia is overcorrection, causing osmotic demyelination syndrome (ODS) — previously called central pontine myelinolysis. When the brain has adapted to chronic hyponatraemia (over >48 hours) by losing osmolytes, rapid sodium correction causes a sudden osmotic shift: water moves out of brain cells, causing demyelination of the pontine white matter. ODS presents 2–4 days after correction with dysarthria, dysphagia, horizontal gaze palsy, quadriplegia, and potentially coma. It is irreversible.
Safe correction limits (to prevent ODS):
- Do NOT exceed 10–12 mmol/L in the first 24 hours and 18 mmol/L in the first 48 hours.
- If correction has been too rapid, STOP sodium supplementation and consider infusing 5% dextrose or desmopressin to re-lower sodium back toward the target — correcting the overcorrection.
Acute severe symptomatic hyponatraemia (seizures, coma): use 3% hypertonic saline 100 mL IV bolus over 10–15 minutes, repeated up to three times; target: raise Na by 4–6 mmol/L rapidly enough to stop symptoms, then maintain safe correction rate. Do NOT give hypertonic saline for chronic asymptomatic hyponatraemia.
Chronic euvolaemic hyponatraemia (SIADH): first-line is fluid restriction to 500–1000 mL/day. If resistant: demeclocycline (blocks ADH effect in collecting duct) or tolvaptan (V2 receptor antagonist — 'vaptans' promote free water excretion without sodium loss, rapidly effective but expensive and requires close monitoring for overcorrection).
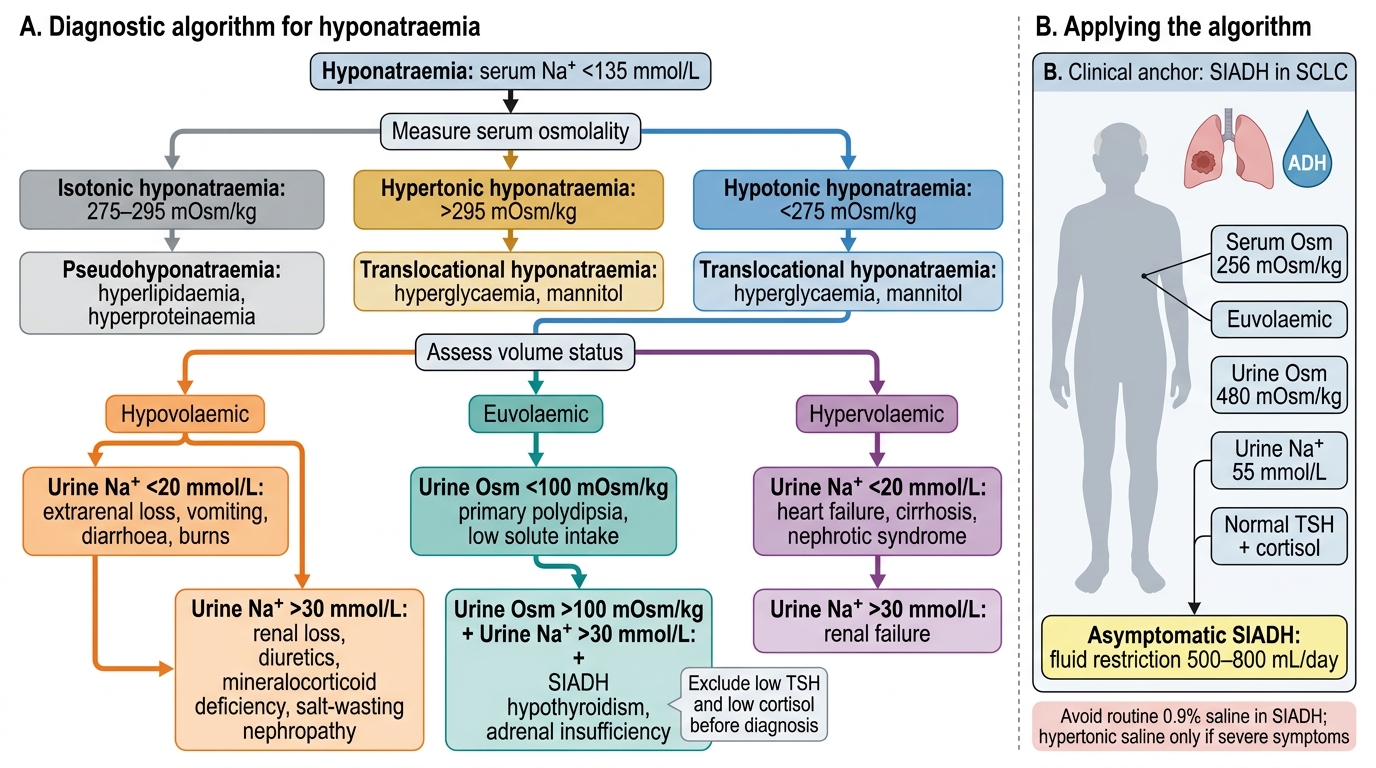
Diagnostic Algorithm for Hyponatraemia
SELF-CHECK
A 68-year-old woman with small cell lung carcinoma is found to have serum sodium of 122 mmol/L. She is clinically euvolaemic, with no oedema and no signs of volume depletion. Serum osmolality is 256 mOsm/kg. Urine osmolality is 480 mOsm/kg. Urine sodium is 55 mmol/L. TSH and morning cortisol are normal. She is asymptomatic. What is the most appropriate first-line management?
A. Intravenous 3% hypertonic saline at 1 mL/kg/hour
B. Intravenous 0.9% normal saline 1 litre over 8 hours
C. Fluid restriction to 500–800 mL per day
D. Oral sodium chloride tablets 3 g three times daily
Reveal Answer
Answer: C. Fluid restriction to 500–800 mL per day
This is classic SIADH (euvolaemic hypotonic hyponatraemia with inappropriately concentrated urine osmolality and elevated urine Na in a patient with SCLC — commonest cause of ectopic ADH). First-line management for asymptomatic SIADH is fluid restriction to 500–1000 mL/day. Normal saline can paradoxically worsen hyponatraemia in SIADH — the sodium is excreted in urine while the water is retained. Hypertonic saline is reserved for acute severe symptomatic hyponatraemia (seizures, coma). Sodium chloride tablets alone are not effective in euvolaemic SIADH without fluid restriction.