Page 8 of 22
OG23.3 | Precocious Puberty — SDL Guide
Learning Objectives
- Define precocious puberty and state the age threshold in girls (<8 years)
- Classify precocious puberty as central (GnRH-dependent) or peripheral (GnRH-independent) and enumerate the causes of each
- Distinguish true precocious puberty from benign variants: premature thelarche and premature adrenarche
- Describe the investigation pathway including GnRH stimulation test, bone age, brain MRI, and pelvic ultrasound
- Outline the principles of management: GnRH analogues for central precocious puberty and cause-directed therapy for peripheral causes
INSTRUCTIONS
A 7-year-old girl presenting with breast buds and pubic hair is one of the most challenging — and consequential — presentations in paediatric and adolescent gynaecology. Misclassifying a benign variant as true precocious puberty leads to unnecessary investigation and treatment. Failing to recognise true precocious puberty allows advancing bone age to silently rob a child of her adult height and exposes her to inappropriate early sexual development. This module provides the framework for accurate classification, efficient investigation, and appropriate management.
References
- DC Dutta's Textbook of Gynecology, 8th ed., Ch. 26 — Disorders of Puberty (textbook)
- Shaw's Textbook of Gynaecology, 17th ed., Ch. 7 — Puberty and Its Disorders (textbook)
- Williams Gynecology, 4th ed., Ch. 14 — Pediatric and Adolescent Gynecology (textbook)
- Carel JC, Léger J. Precocious puberty. NEJM 2008;358:2366-2377 (guideline)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A mother brings her 6-year-old daughter, noting that she has developed breast buds over the past three months and has started growing much faster than classmates. Bone age X-ray shows a skeletal age of 9 years. The child is otherwise well. What is the diagnosis? What single investigation will most efficiently tell you whether this child needs treatment — and what is at stake if she does not receive it?
WHY THIS MATTERS
Precocious puberty affects approximately 1 in 5,000–10,000 girls. Its consequences extend well beyond early physical development: without treatment, the premature growth spurt followed by early epiphyseal fusion leads to short adult stature — a girl who starts puberty at age 6 may stop growing at age 10–11 and reach only 150–155 cm, losing 5–10 cm of potential height. There are also significant psychosocial consequences — early puberty in a 6–7-year-old exposes a child to menstruation, body hair, and secondary sex characteristics that she and her peers are unprepared for. For the clinician, the key skill is accurate classification: the majority of central precocious puberty in girls is idiopathic and responds well to GnRH analogue treatment, but a subset of cases signals an intracranial tumour or adrenal pathology that must not be missed.
RECALL
Recall from the previous modules: normal puberty in girls begins at age 8–13. The HPO axis is activated by GnRH pulsatility from KNDy neurons in the arcuate nucleus, driving LH and FSH secretion. LH and FSH stimulate the ovary to produce oestradiol. In adrenarche, the adrenal zona reticularis independently secretes DHEAS and androstenedione, driving pubic and axillary hair. These two events — gonadarche (HPO axis) and adrenarche (adrenal) — are independent and can occur at different times. Precocious puberty arises from premature activation of either the HPO axis (central) or autonomous sex steroid production (peripheral).
Clinical Presentation of Precocious Puberty
Precocious puberty is defined as the development of secondary sexual characteristics before the lower limit of the normal age range: before age 8 years in girls and before age 9 years in boys. The earlier and more rapidly puberty progresses, the more likely an underlying pathological cause — although in girls, the majority of central precocious puberty remains idiopathic even after full investigation.
The clinical presentation varies by the type of precocious puberty and the specific cause, but the common presenting features include breast development in a girl younger than 8, pubic or axillary hair in a young child, accelerated growth velocity that places the child significantly above her expected height centile for age, and — in more advanced cases — vaginal discharge or bleeding. The parent typically notices that the child is growing faster than peers, developing a body odour, and showing physical changes that are unexpected at her age. Acne and body odour are driven by adrenal androgens and may precede or accompany other signs.
Clinically, the examination must establish: (a) the exact Tanner stage for breast and pubic hair — documenting precisely what is present and at what stage; (b) the growth velocity — has there been an acceleration? The height centile chart is an invaluable tool; (c) neurological signs — headache, visual field defects, and gelastic (laughing) seizures raise the possibility of intracranial pathology; (d) skin findings — café-au-lait patches with irregular ('coast of Maine') borders suggest McCune-Albright syndrome (peripheral); smooth-bordered café-au-lait spots suggest neurofibromatosis type 1. An important diagnostic distinction must be made between true (complete) precocious puberty, in which multiple pubertal signs are progressing in sequence, and benign variants such as isolated premature thelarche or isolated premature adrenarche, in which only one sign is present in isolation without the others.
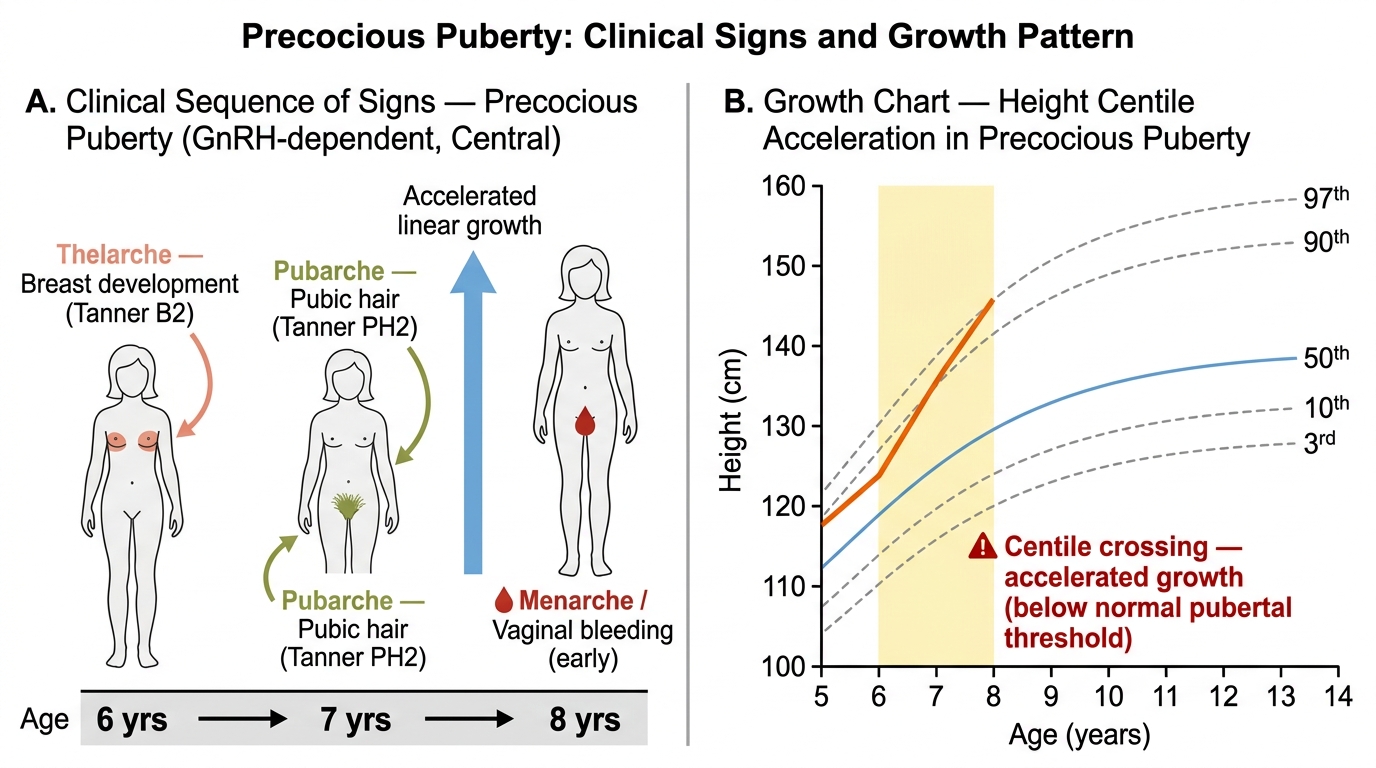
Precocious Puberty: Sequence of Clinical Signs and Growth Chart Acceleration (Ages 6–8)
Pathophysiology and Causes of Precocious Puberty
The classification of precocious puberty into central and peripheral types is based on whether the HPO axis is activated (central, GnRH-dependent) or whether sex steroids are produced independently of the HPO axis (peripheral, GnRH-independent). This distinction is clinically critical because it determines the entire investigation and management pathway. The most important single test that bifurcates these two categories is the measurement of LH and FSH — elevated gonadotrophins point to central activation, while suppressed gonadotrophins in the presence of elevated sex steroids point to peripheral autonomous production. Understanding the mechanism behind each cause allows the clinician to predict the gonadotrophin pattern and select the correct investigation.
Central (GnRH-dependent) Precocious Puberty:
The entire HPO axis is prematurely activated. GnRH is secreted in pulses, LH and FSH rise to pubertal levels, and the ovary responds with oestradiol production and follicular development. The sequence of pubertal events is normal (thelarche → pubarche → menarche) but compressed and advanced in time.
- Idiopathic central precocious puberty: Accounts for approximately 80–90% of central precocious puberty in girls. No identifiable cause is found despite full investigation. It is far less common for idiopathic central PP to occur in boys (~25%), which is why boys with central PP require more aggressive CNS investigation.
- Hypothalamic hamartoma: The most common pathological cause of central PP. A benign ectopic mass of GnRH-secreting hypothalamic tissue that acts as an ectopic GnRH pulse generator, independently of the normal pubertal timing mechanisms. Characteristically associated with gelastic seizures (brief, inappropriate laughing episodes). Visible on MRI as a sessile or pedunculated mass attached to the hypothalamus.
- Other CNS causes: Optic glioma (associated with neurofibromatosis type 1), craniopharyngioma (hypothalamic compression), astrocytoma, ependymoma, CNS irradiation, hydrocephalus, and prior meningitis/encephalitis. All cause central precocious puberty by disrupting the normal central inhibitory control of GnRH secretion.
Peripheral (GnRH-independent) Precocious Puberty:
Sex steroids are produced autonomously, bypassing the normal HPO regulatory axis. The feedback of high oestrogen (or androgens) suppresses GnRH pulsatility, resulting in LOW LH and FSH — the opposite of central PP. This is the key diagnostic discriminator.
- McCune-Albright syndrome: A sporadic activating mutation in the GNAS1 gene (encoding Gs-alpha protein) causing autonomous sex steroid production from ovarian cysts. Classic triad: polyostotic fibrous dysplasia (bone lesions causing deformity and fractures), café-au-lait skin patches (large, irregular 'coast of Maine' borders, often unilateral), and autonomous ovarian cysts (causing episodic oestrogenisation — vaginal bleeding may precede breast development in a pattern unlike normal puberty). Ovarian ultrasound shows large follicular cysts.
- Granulosa cell tumour of the ovary: An oestrogen-secreting sex-cord stromal tumour; one of the most common ovarian tumours in prepubertal girls. Presents with signs of oestrogen excess (breast development, vaginal bleeding, growth acceleration); LH/FSH are suppressed. Inhibin B is elevated (tumour marker). Management is surgical — unilateral oophorectomy is usually curative.
- Congenital adrenal hyperplasia (CAH): 21-hydroxylase deficiency leads to cortisol deficiency and massive ACTH-driven adrenal androgen overproduction (17-OHP elevated). Androgens drive premature adrenarche and accelerated bone age. In girls, virilisation (clitoral hypertrophy) may be present. This is classified as peripheral PP because the gonadotrophin axis is not driving the changes.
- Exogenous sex steroids: Accidental or deliberate exposure to oestrogen-containing creams, contraceptive pills, or phytoestrogens. History is critical.
Benign Variants (NOT true precocious puberty):
- Premature thelarche: Isolated breast budding before age 8, with prepubertal LH/FSH, normal bone age, and no other pubertal signs. Usually occurs between ages 1–3 and is self-limiting. Requires monitoring to confirm it does not progress to true precocious puberty (repeat Tanner staging and bone age in 6 months).
- Premature adrenarche: Isolated pubic/axillary hair before age 8, driven by early adrenal DHEAS production. LH/FSH prepubertal; 17-OHP normal (to exclude CAH). Benign, but associated with later PCOS and metabolic syndrome — counsel accordingly.
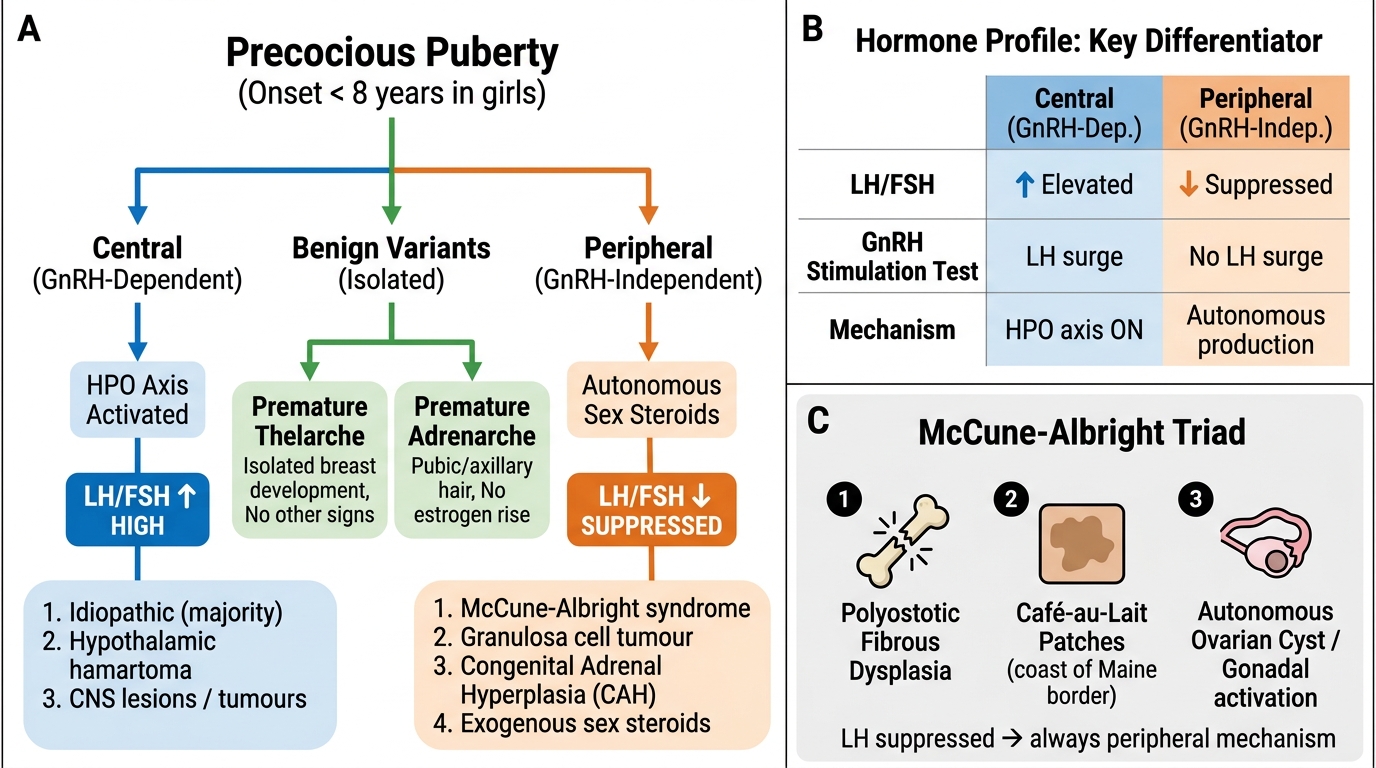
Classification of Precocious Puberty: Central vs Peripheral vs Benign Variants
SELF-CHECK
A 7-year-old girl has breast development and pubic hair. Her LH is suppressed at 0.1 IU/L. Oestradiol is elevated. Pelvic ultrasound shows a 4 cm ovarian cyst. She has large café-au-lait patches with irregular borders on her left flank and an X-ray shows bony lesions in her left femur. The most likely diagnosis is:
A. Idiopathic central precocious puberty
B. Hypothalamic hamartoma with central precocious puberty
C. McCune-Albright syndrome (peripheral precocious puberty)
D. Granulosa cell tumour of the ovary
Reveal Answer
Answer: C. McCune-Albright syndrome (peripheral precocious puberty)
The classic triad of McCune-Albright syndrome is present: polyostotic fibrous dysplasia (bony lesions), café-au-lait patches with irregular 'coast of Maine' borders, and autonomous ovarian cyst causing oestrogen excess. Crucially, LH is SUPPRESSED — confirming GnRH-independent (peripheral) mechanism. Central causes would have elevated LH/FSH. Granulosa cell tumour also suppresses LH but lacks the skin and bone findings.
Diagnosis and Investigation of Precocious Puberty
Investigation of precocious puberty follows a systematic pathway guided by the clinical presentation and the central vs peripheral classification. The initial approach distinguishes true precocious puberty (multiple progressing pubertal signs, accelerated bone age) from benign variants (isolated single sign, normal bone age), because benign variants need only monitoring and not full endocrine investigation. When the presentation suggests true precocious puberty, the investigation pathway proceeds in stages, with the GnRH stimulation test serving as the critical bifurcation test between central and peripheral disease.
Before ordering specialist investigations, a thorough clinical history and examination should identify: (a) the rate of progression — is puberty advancing rapidly over weeks, or has it been static for months? Static findings over 6 months are more consistent with a benign variant; (b) any neurological symptoms (headache, visual changes, seizures) suggesting a CNS cause; (c) skin findings (café-au-lait patches) suggesting McCune-Albright or NF1; (d) abdominal examination — an ovarian or adrenal mass may be palpable.
The investigation pathway is as follows:
All cases with true precocious puberty (multiple progressing signs):
- Bone age X-ray (left wrist): Advanced bone age (>2 SD above mean for chronological age) confirms that the skeleton is being driven by sex steroids; this is both diagnostic support and a baseline for monitoring treatment response.
- Basal LH and FSH: The pivotal first test. Prepubertal: LH <0.1–0.3 IU/L; pubertal: LH ≥0.3–1 IU/L. Suppressed LH with elevated oestradiol = peripheral PP.
- GnRH (or GnRH agonist) stimulation test: The gold standard for distinguishing central from peripheral PP. A pubertal LH response (peak LH >5–8 IU/L, with LH:FSH ratio >1) confirms central HPO axis activation. A flat or suppressed LH response confirms peripheral (autonomous) PP.
- Oestradiol and/or testosterone (if virilisation present).
- Pelvic ultrasound: Ovarian volume, follicle size, and uterine size. In central PP, uterine length >3.5 cm and ovarian volumes >2 mL support HPO activation. An ovarian cyst (McCune-Albright) or solid mass (granulosa cell tumour) indicates peripheral cause.
- Adrenal steroid profile (17-OHP, DHEAS, cortisol): If virilisation or clinical suspicion of CAH.
If central PP confirmed:
- MRI brain (hypothalamus and pituitary): Mandatory to exclude intracranial pathology — hypothalamic hamartoma, craniopharyngioma, optic glioma. Even in girls where the majority of cases are idiopathic, MRI is essential because missing a treatable tumour has serious consequences. In girls aged 6–8 with central PP and no neurological symptoms, MRI findings are abnormal in approximately 10–15% of cases.
If peripheral PP confirmed:
- Pelvic MRI: Ovarian lesion characterisation.
- Inhibin B and AMH: Elevated in granulosa cell tumour.
- Skeletal survey: Polyostotic fibrous dysplasia in McCune-Albright.
- Urinary/serum steroid profile: CAH confirmation (17-OHP, ACTH stimulation test).
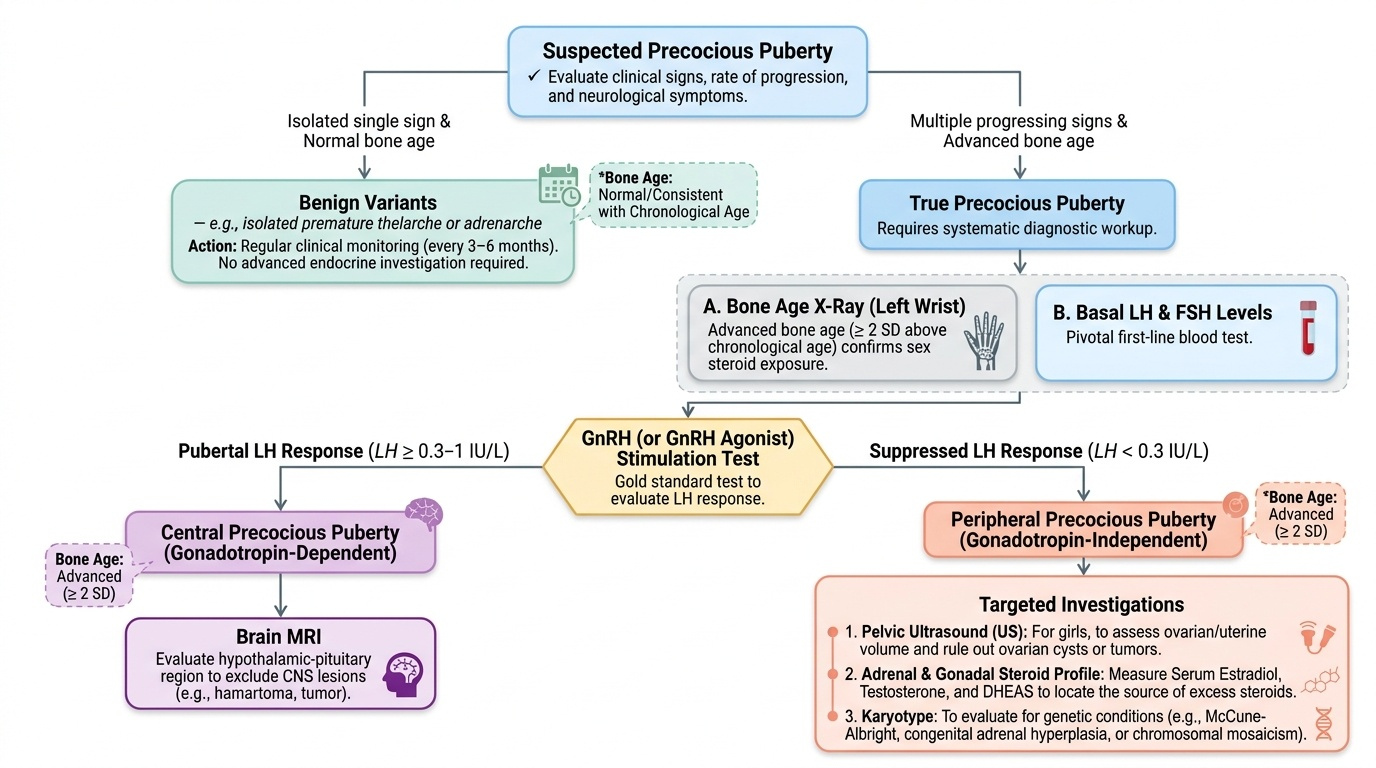
Provided image
SELF-CHECK
The GnRH stimulation test is the gold standard for distinguishing central from peripheral precocious puberty. In central (GnRH-dependent) precocious puberty, the expected LH response to GnRH stimulation is:
A. LH remains suppressed (flat response); FSH rises
B. LH rises to pubertal range (peak >5–8 IU/L) with LH:FSH ratio >1
C. LH and FSH both remain suppressed
D. LH rises markedly but oestradiol falls
Reveal Answer
Answer: B. LH rises to pubertal range (peak >5–8 IU/L) with LH:FSH ratio >1
In central (GnRH-dependent) precocious puberty, the pituitary has been sensitised by chronic GnRH pulsatility and responds to exogenous GnRH with a pubertal LH surge (peak LH >5–8 IU/L), with LH:FSH ratio >1 (LH-dominant response, reflecting luteinising cell maturation). In peripheral PP, the pituitary is suppressed by chronic high sex steroids and the LH response is flat. This is why GnRH stimulation is the bifurcating test.