Page 22 of 29
EN4.20 | Deaf Child — SDL Guide
Learning Objectives
- Describe the clinical features and developmental milestones relevant to identifying hearing loss in a child
- Classify childhood hearing loss by type, severity, onset, and aetiology
- Describe the genetic and acquired causes of SNHL in children, including connexin 26 and TORCH infections
- Outline the investigation pathway including universal neonatal screening (OAE and ABR)
- Describe the management principles including early hearing aid fitting, cochlear implantation eligibility and timing, and the critical period for language development
INSTRUCTIONS
Childhood hearing loss is a critical topic because intervention before the critical period for speech and language development (the first 2–3 years of life) dramatically improves language outcomes. Missing or delaying diagnosis has lifelong consequences. EN4.20 requires you to describe clinical features, investigations, and management of the deaf child, including the role of cochlear implantation.
References
- Dhingra PL — Diseases of Ear, Nose and Throat, 7th ed., Ch. 5 (Deafness in Children) (textbook)
- Hazarika P — Textbook of Ear Nose Throat and Head & Neck Surgery, 3rd ed. (textbook)
- Scott-Brown's Otorhinolaryngology, Head and Neck Surgery, 8th ed., Vol. 3, Ch. 236 (textbook)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A 9-month-old boy is brought to the paediatric outpatient by his mother, who is concerned that 'he doesn't respond to my voice.' He failed his OAE hearing screen at birth but the family did not follow up as advised. He shows no startle response to loud sounds, does not babble, and makes only undifferentiated vowel sounds. His 3-year-old sister has normal hearing. What is the most likely diagnosis, what has been lost by the 9-month delay in following up, and what urgent investigation and intervention are needed?
WHY THIS MATTERS
Hearing loss is the most common congenital sensory disability in children, affecting approximately 1–2 per 1000 live births with severe-to-profound bilateral SNHL, and a further 2–3 per 1000 with moderate bilateral hearing loss. In India, the scale is enormous: with approximately 27 million births per year, approximately 27,000–54,000 children are born each year with significant hearing loss — many in resource-limited settings without access to universal screening. The consequences of undetected childhood hearing loss are profound: language acquisition, speech development, literacy, academic achievement, and social development all depend on auditory input during the critical period of the first 2–3 years. A child who is born with profound bilateral SNHL and is not identified and fitted with hearing aids or implanted with a cochlear implant before age 2–3 will never achieve age-appropriate spoken language, even with intervention later in life. The 1-3-6 month rule — diagnose by 1 month, confirm by 3 months, intervene by 6 months — represents the global standard for neonatal hearing screening programmes, and the deviation from this rule carries a compounding developmental cost.
RECALL
Recall the developmental milestones relevant to hearing in a child: by 1 month, a neonate should startle to loud sounds; by 3–4 months, should quieten or smile to a familiar voice; by 6 months, should turn toward sounds; by 9 months, should respond to name; by 12 months, should babble with varied consonant-vowel combinations and say 1–2 words; by 18 months, should have 10–20 words; by 24 months, should combine two words into short phrases. Failure to meet these milestones, particularly the absence of babbling by 9 months or failure to use single words by 12 months, should immediately trigger formal hearing assessment. Recall also that the primary auditory cortex undergoes its most rapid synaptic maturation during the first 2 years — this is the neurological basis for the critical period, and it explains why cochlear implants deliver far better language outcomes when fitted before this window closes.
Clinical Presentation: The Deaf Child
The clinical recognition of hearing loss in a child depends fundamentally on whether the child has been detected through a universal neonatal screening programme or presents later through parental concern or developmental failure. Both pathways are clinically important — universal screening has dramatically reduced the age of diagnosis in countries where it is implemented, but in many Indian settings, presentation is still through parental concern, often after months of delay.
In the neonatal period, a failed otoacoustic emission (OAE) screen prompts referral for ABR (auditory brainstem response) testing. A failed ABR confirms the need for formal audiological assessment and aetiology investigation. The first 9 months of life are the critical window — an infant referred at birth who is not followed up until 9 months (as in the opening scenario) has lost half the critical language-development period without any auditory input.
In older infants and children who present through parental concern, the clinical features depend on the degree of hearing loss and the child's age. Clues from the history and developmental assessment include absence of startle response to loud sounds in early infancy, failure to turn toward sounds by 6 months, failure to babble by 9–12 months, reduced or absent single words by 12–15 months, and absent two-word combinations by 24 months. Parents often report that the child 'responds inconsistently' — sometimes responding and sometimes not, which reflects the fluctuating nature of some CHL (e.g. glue ear) or the child's use of vibrotactile cues and lip-reading to compensate.
Key parental concerns that should always trigger formal audiological referral:
- Any parental concern about hearing
- Failed neonatal screen not followed up
- Delayed speech or language milestones
- The child seems to 'hear selectively' or ignores instructions
- The child watches the speaker's face intently (lip-reading)
- Increasing volume of television or toys
- The child has had meningitis, severe prematurity, or is on/was on ototoxic medication
Aetiology and Pathophysiology
The aetiology of childhood hearing loss is heterogeneous — approximately 50% of congenital SNHL is genetic, and 50% is acquired (environmental causes including infections, ototoxic drugs, and perinatal complications). Correctly identifying the aetiology is important not only for prognosis and management planning but also for genetic counselling (if genetic causes are confirmed), identifying associated systemic abnormalities in syndromic conditions, and selecting appropriate treatment for acquired causes. The aetiology also determines the urgency of intervention: post-meningitic cochlear ossification is a surgical emergency, congenital CMV detected within 1 month of birth can be treated with antiviral therapy to slow progression, and syndromic causes require multidisciplinary assessment of other organ systems. The following categorisation of causes provides the framework for the aetiological investigation protocol.
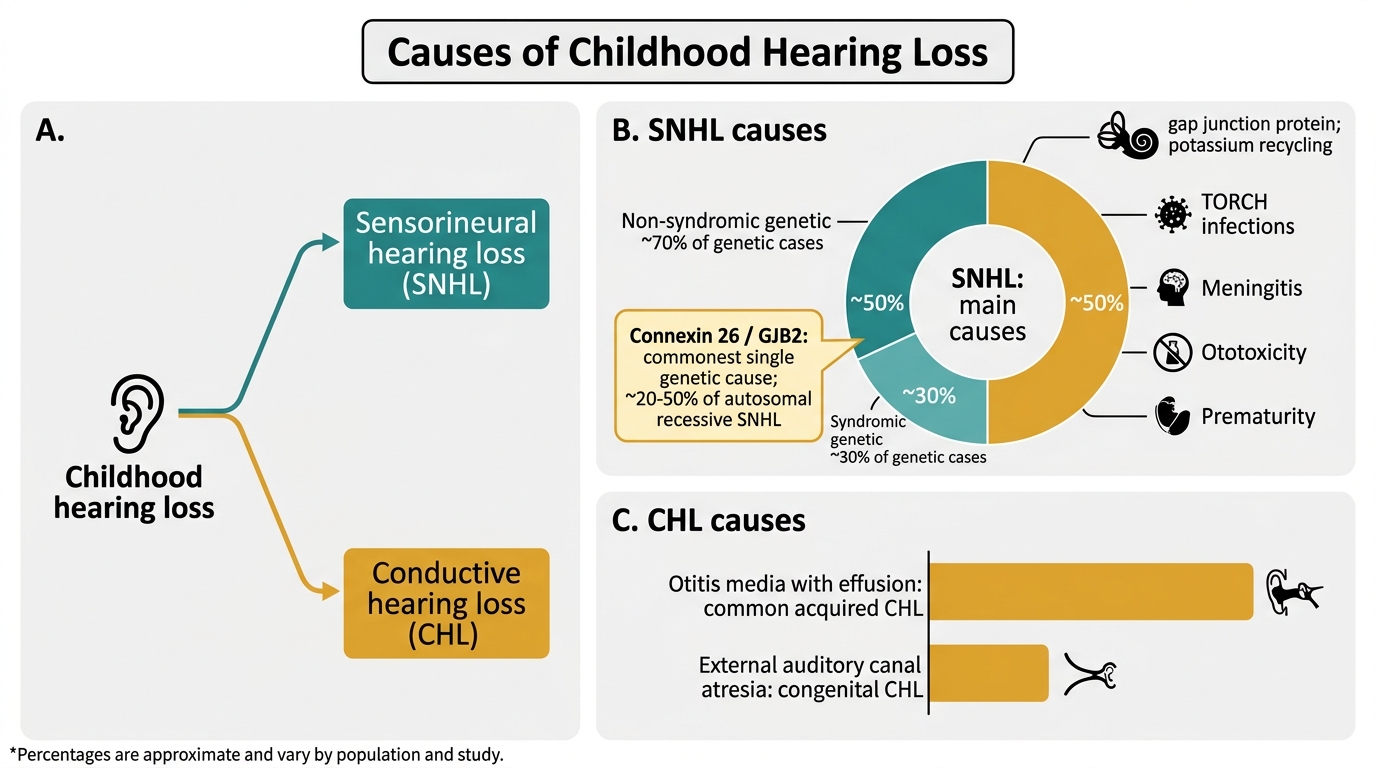
Causes of Childhood Hearing Loss
Genetic causes (~50% of SNHL):
Genetic hearing loss is divided into non-syndromic (hearing loss without other abnormalities, ~70% of genetic cases) and syndromic (hearing loss with other organ involvement, ~30%). The most important non-syndromic cause is connexin 26 mutation (GJB2 gene, chromosome 13q11) — mutations in this gene account for approximately 20–50% of autosomal recessive SNHL, making it the most common single genetic cause of childhood deafness. Connexin 26 encodes a gap junction protein in the cochlear supporting cells critical for potassium ion recycling in the endolymph.
Key syndromic causes:
- Waardenburg syndrome (autosomal dominant, PAX3 gene): white forelock, premature greying, telecanthus (dystopia canthorum), heterochromia iridis, + bilateral SNHL. Type I and II differ by dystopia canthorum.
- Usher syndrome (autosomal recessive): SNHL + progressive retinitis pigmentosa (later visual field loss and night blindness); the most common cause of combined deaf-blindness
- Pendred syndrome (autosomal recessive, SLC26A4/pendrin gene): bilateral SNHL + goitre (thyroid enlargement); enlarged vestibular aqueduct on CT temporal bone
- Alport syndrome (X-linked, COL4A5 gene): progressive SNHL + glomerulonephritis (haematuria, proteinuria) + ocular abnormalities (lenticonus)
Acquired causes (~50%):
- TORCH infections (Toxoplasma, Rubella, Cytomegalovirus, Herpes simplex): congenital infection damages the developing cochlea. Congenital CMV is now the most common non-genetic cause of congenital SNHL in vaccinated populations; it may be asymptomatic at birth but cause progressive, sometimes late-onset SNHL; treated with valganciclovir if hearing loss detected within 1 month of birth.
- Meningitis (bacterial — Haemophilus, Pneumococcus, Meningococcus): cochlear ossification (labyrinthitis ossificans) can occur rapidly after bacterial meningitis, making early implantation urgent in children with post-meningitic profound SNHL before the cochlea ossifies.
- Perinatal factors: prematurity (<32 weeks), very low birth weight, hypoxia, hyperbilirubinaemia (kernicterus), neonatal sepsis.
- Ototoxic drugs: gentamicin (common in neonatal ICU settings in India), furosemide, cisplatin.
- Otitis media with effusion (glue ear): the most common cause of CHL in children aged 2–7 years; bilateral CHL of 20–40 dB; managed conservatively in most cases.
SELF-CHECK
A 2-year-old boy is found on audiometry to have bilateral profound SNHL. Examination shows a white forelock and heterochromia iridis. His father has grey hair at age 35. The most likely genetic diagnosis is:
A. Usher syndrome (SNHL + retinitis pigmentosa)
B. Waardenburg syndrome (SNHL + pigmentary abnormalities + autosomal dominant)
C. Pendred syndrome (SNHL + goitre)
D. Alport syndrome (SNHL + nephritis)
Reveal Answer
Answer: B. Waardenburg syndrome (SNHL + pigmentary abnormalities + autosomal dominant)
The combination of bilateral SNHL, white forelock, heterochromia iridis, and autosomal dominant family history (grey hair in father at 35) is pathognomonic of Waardenburg syndrome. Usher syndrome causes SNHL + retinitis pigmentosa, not pigmentary abnormalities of the hair or iris. Pendred syndrome causes SNHL + goitre. Alport syndrome causes SNHL + nephritis. Waardenburg syndrome is caused by PAX3 gene mutations and is the most common syndromic cause of autosomal dominant SNHL.
Investigations
The investigation of childhood hearing loss follows a systematic pathway from population-level screening to individual diagnostic assessment, then aetiological investigation. Each step uses age-appropriate tests because children cannot cooperate with standard adult audiometry until approximately age 5–6 years. Understanding which test is used at which age, and what it measures, is clinically important. The paediatric audiological assessment pathway is structured around the physiological maturity of the child's auditory and behavioural systems: in the newborn and early infant, only electrophysiological tests are possible; from 6 months, conditioned behavioural responses can be used; from 2.5 years, play audiometry works; and from 5–6 years, standard pure-tone audiometry becomes reliable. The table below summarises these age-appropriate audiological tools and the transition points between them.
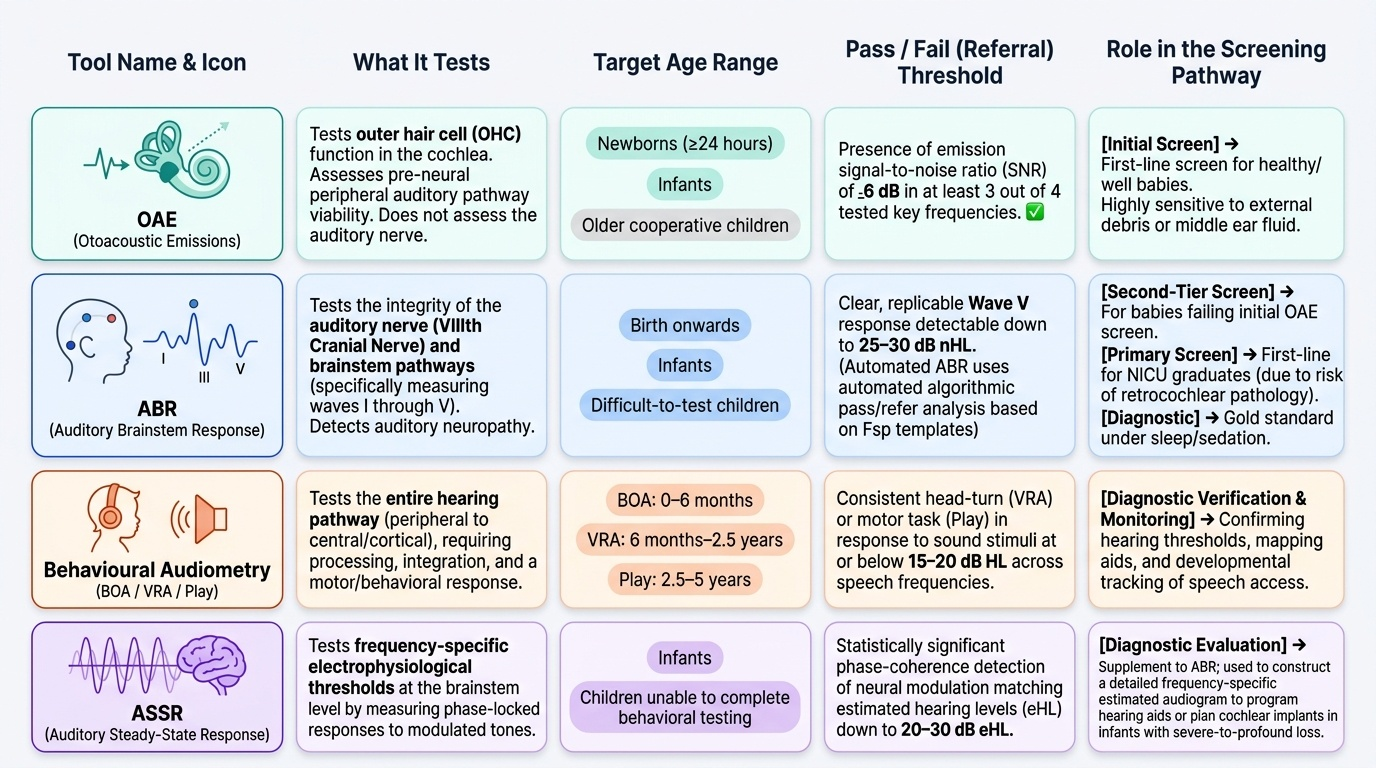
Provided image
1. Otoacoustic emissions (OAEs): A fast, non-invasive screen for cochlear outer hair cell function. Used as the first-line neonatal screen (by 1 month of age). A pass result indicates outer hair cells are functioning; a fail requires ABR. OAEs detect cochlear SNHL but cannot detect neural hearing loss (auditory neuropathy spectrum disorder, ANSD).
2. Auditory brainstem response (ABR): Electrophysiological measurement of VIII nerve and brainstem auditory pathway. Provides objective threshold estimation; not dependent on behavioural cooperation. Used when OAE fails or to confirm SNHL; can diagnose auditory neuropathy. ABR threshold >35 dB HL = refer for formal audiological assessment. Can be performed under natural sleep in infants or sedation.
3. Auditory steady-state response (ASSR): A frequency-specific electrophysiological threshold test; used to programme hearing aids and cochlear implants in infants; can estimate hearing thresholds at multiple frequencies simultaneously.
4. Behavioural audiometry (age-dependent):
- Visual reinforcement audiometry (VRA, 6 months – 2.5 years): conditioned head-turning toward a sound, rewarded with a visual stimulus.
- Play audiometry (2.5–5 years): child performs a task in response to a sound.
- Standard pure-tone audiometry (>5 years): conventional audiogram.
5. Tympanometry: Essential for detecting CHL (middle ear effusion, ossicular problems). Type B tympanogram (flat) = effusion.
6. CT temporal bone / MRI inner ear:
HRCT: cochlear abnormalities (Mondini deformity, cochlear aplasia), ossification (post-meningitic), enlarged vestibular aqueduct (Pendred). MRI: VIII nerve anatomy (aplasia, stenosis — critical before cochlear implantation); cochlear fluid spaces.
7. Aetiological investigations:
- TORCH serology (maternal and neonatal)
- CMV urine PCR (in infants — most sensitive within 3 weeks of birth)
- Genetic testing: GJB2/GJB6 (connexin 26/30) mutation analysis; chromosomal microarray; next-generation sequencing panels for deafness genes
- Ophthalmology (retinal examination for Usher syndrome, Alport eye signs)
- Renal ultrasound + urinalysis (Alport syndrome)
- Thyroid function (Pendred syndrome)