Page 16 of 30
PE20.6 | Chronic Renal Failure — SDL Guide
Learning Objectives
- Recognise the insidious clinical presentation of chronic kidney disease (CKD) in children including growth failure, anaemia, and hypertension
- Classify paediatric CKD by aetiology (CAKUT, glomerular, hereditary) and stage by KDIGO eGFR criteria G1–G5
- Enumerate the major complications of CKD: renal osteodystrophy, anaemia, growth failure, hypertension, and electrolyte disturbances
- Plan management strategies including slowing CKD progression (RAS blockade), treating each complication, and indications for renal replacement therapy
- Counsel families on prognosis and the importance of long-term monitoring
INSTRUCTIONS
Unlike acute kidney injury, chronic kidney disease in children develops silently over months to years and is often discovered late — sometimes when a child is already severely uraemic. The two unique dimensions of paediatric CKD that distinguish it from adult disease are its impact on growth and skeletal development during a critical window, and the predominance of congenital/structural causes rather than the diabetes and hypertension that drive adult CKD. This module covers the full arc from presentation through pathophysiology, staging, complication management, and renal replacement planning.
References
- Ghai Essential Pediatrics, 9th ed., Ch. 18 — Kidney Diseases (textbook)
- Nelson Textbook of Pediatrics, 21st ed., Ch. 553 — Chronic Kidney Disease (textbook)
- KDIGO 2012 Clinical Practice Guideline for the Evaluation and Management of CKD (guideline)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A 9-year-old girl is referred for evaluation of short stature. She is below the 3rd percentile for height and weight, pale, and her mother mentions she often wakes at night to pass urine. Blood pressure is 130/86 mmHg. Her blood tests show creatinine 2.4 mg/dL, haemoglobin 7.8 g/dL, calcium 7.8 mg/dL, phosphate 6.2 mg/dL, and PTH 340 pg/mL. Renal ultrasound shows bilateral small, echogenic kidneys. How did she get here, and what are your priorities?
WHY THIS MATTERS
Chronic kidney disease is one of the most demanding conditions in paediatric medicine because it is irreversible, progressive, and affects not only renal function but the entire trajectory of a child's physical development. In India, CKD is detected late in many children because the early symptoms — mild growth faltering, nocturia, subtle anaemia — are easily attributed to nutritional deficiency or other common conditions. Final-year students entering paediatric postings will encounter children with CKD at multiple stages, from an incidentally detected proteinuric child to a teenager requiring dialysis. Understanding the staging system, the major complications, and the management framework equips you to act promptly and counsel families accurately on what lies ahead.
RECALL
Before proceeding, recall the following foundational concepts:
- GFR and eGFR: the Schwartz formula estimates GFR in children using height and serum creatinine (eGFR = k × height in cm / creatinine in mg/dL; k=0.413 for most children). Normal GFR in children >2 years ≈ 90–120 mL/min/1.73m².
- PTH–calcium–phosphate axis: the kidney activates vitamin D (25(OH)D → 1,25(OH)2D3) and excretes phosphate. Renal failure → low vitamin D activation + phosphate retention → hypocalcaemia → secondary hyperparathyroidism.
- Erythropoietin: produced by peritubular fibroblasts in the kidney; drives red cell production. Destroyed nephrons produce less erythropoietin → normocytic normochromic anaemia.
- Growth hormone axis: GH is secreted by the anterior pituitary; its anabolic effects in uraemia are blunted by GH resistance and poor nutrition, causing profound growth failure in CKD children.
- Holliday-Segar maintenance fluids — baseline for all fluid prescription in paediatric renal disease.
Clinical Presentation of CKD in Children
Chronic kidney disease (CKD) in children rarely announces itself dramatically. Unlike acute kidney injury — which presents over hours to days with a clear precipitant — CKD evolves insidiously over months to years, with the child and family often adapting imperceptibly to the gradually worsening symptoms. Most children are diagnosed either incidentally (on routine urinalysis or imaging), or after presenting with seemingly unrelated complaints such as poor growth, pallor, or recurrent headaches that, on systematic investigation, reveal months to years of progressive renal dysfunction. The insidious onset is one of the greatest diagnostic challenges of paediatric nephrology and explains why many Indian children reach the nephrologist only when CKD is already at G4 or G5.
The clinical presentation of paediatric CKD reflects its systemic consequences:
- Growth failure is often the most prominent sign in children with long-standing CKD. The child falls progressively below the expected growth centile — frequently interpreted as nutritional failure or constitutional short stature before the renal cause is found. In retrospect, growth charts show a gradually widening deficit beginning years before diagnosis.
- Pallor and fatigue from normochromic normocytic anaemia due to erythropoietin deficiency; exercise intolerance and poor school performance are common parental complaints.
- Polyuria and nocturia reflect impaired urinary concentrating ability; the child who begins wetting the bed again at age 8 may have CKD rather than a behavioural problem.
- Hypertension — often asymptomatic but sometimes presenting with headache, epistaxis, or, in severe cases, hypertensive retinopathy or encephalopathy.
- Bone pain and deformities from renal osteodystrophy; rachitic rosary, wrist widening, or genu valgum in younger children.
- Anorexia, nausea, and lethargy are features of established uraemia, typically in advanced (G4–G5) CKD.
- Oedema and nephrotic features when glomerular disease is the cause.
Important historical clues pointing to a congenital/structural cause (CAKUT) include: recurrent urinary tract infections in infancy, antenatal hydronephrosis detected on fetal anomaly scan, known posterior urethral valves or vesicoureteric reflux, and a family history of renal disease or deafness (Alport syndrome). The absence of acute illness, the presence of small echogenic kidneys on ultrasound, and a bilateral symmetrical process all favour CKD over acute injury.
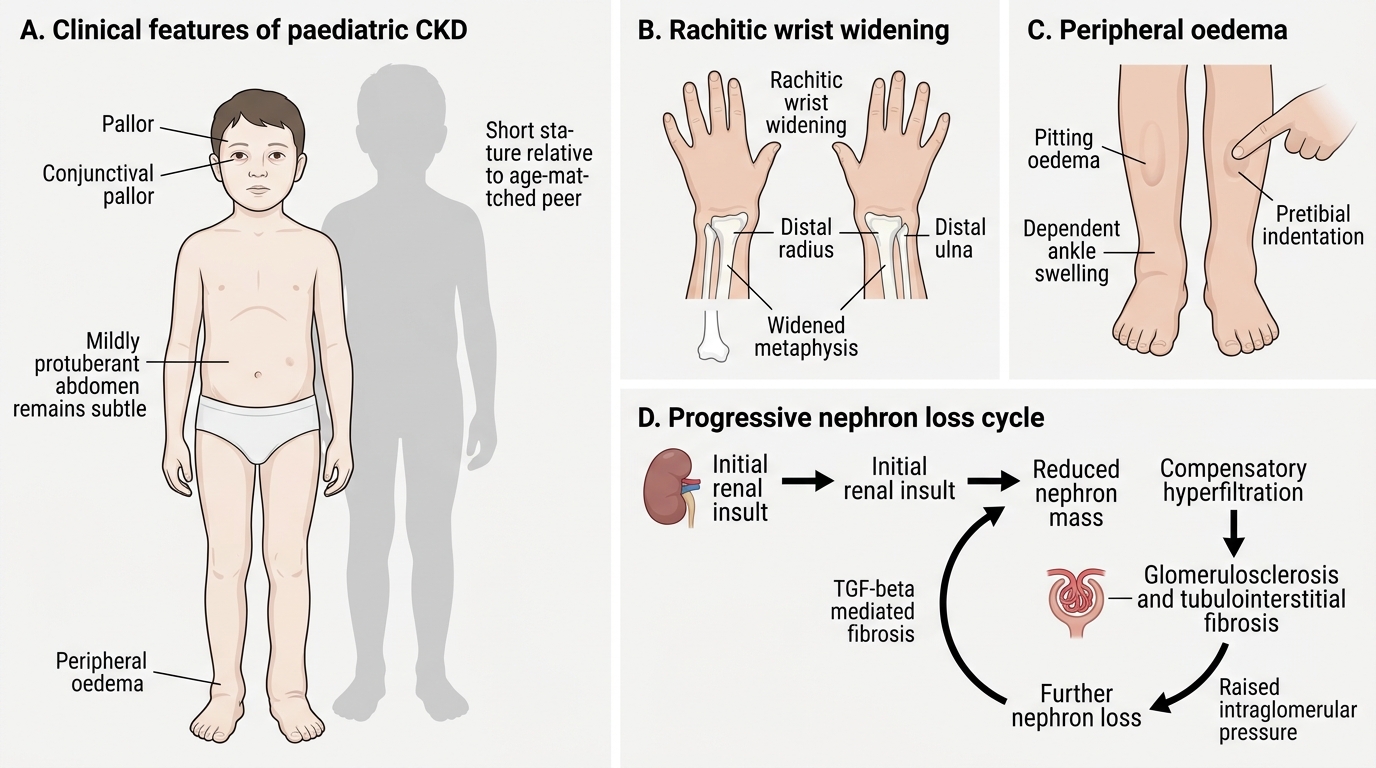
Clinical Features and Progression of Paediatric CKD
Pathophysiology and Aetiology of Paediatric CKD
Understanding why paediatric CKD progresses even after the original insult is removed is essential for rational management. The progression is driven by a self-amplifying cycle of nephron loss, compensatory hyperfiltration, and subsequent scarring that continues even when the primary disease is quiescent.
Progressive nephron loss — the final common pathway:
When enough nephrons are destroyed by any cause, the remaining nephrons undergo compensatory hypertrophy and hyperfiltration to maintain adequate GFR. This adaptive response is ultimately maladaptive: the elevated intraglomerular pressure causes glomerular endothelial injury, promotes mesangial expansion, and triggers TGF-β–mediated tubulointerstitial fibrosis. The result is progressive glomerulosclerosis and further nephron loss — a vicious cycle that perpetuates regardless of whether the original disease is still active. This is why blood pressure control and proteinuria reduction (via RAS blockade) are the most evidence-based interventions for slowing CKD progression in any cause.
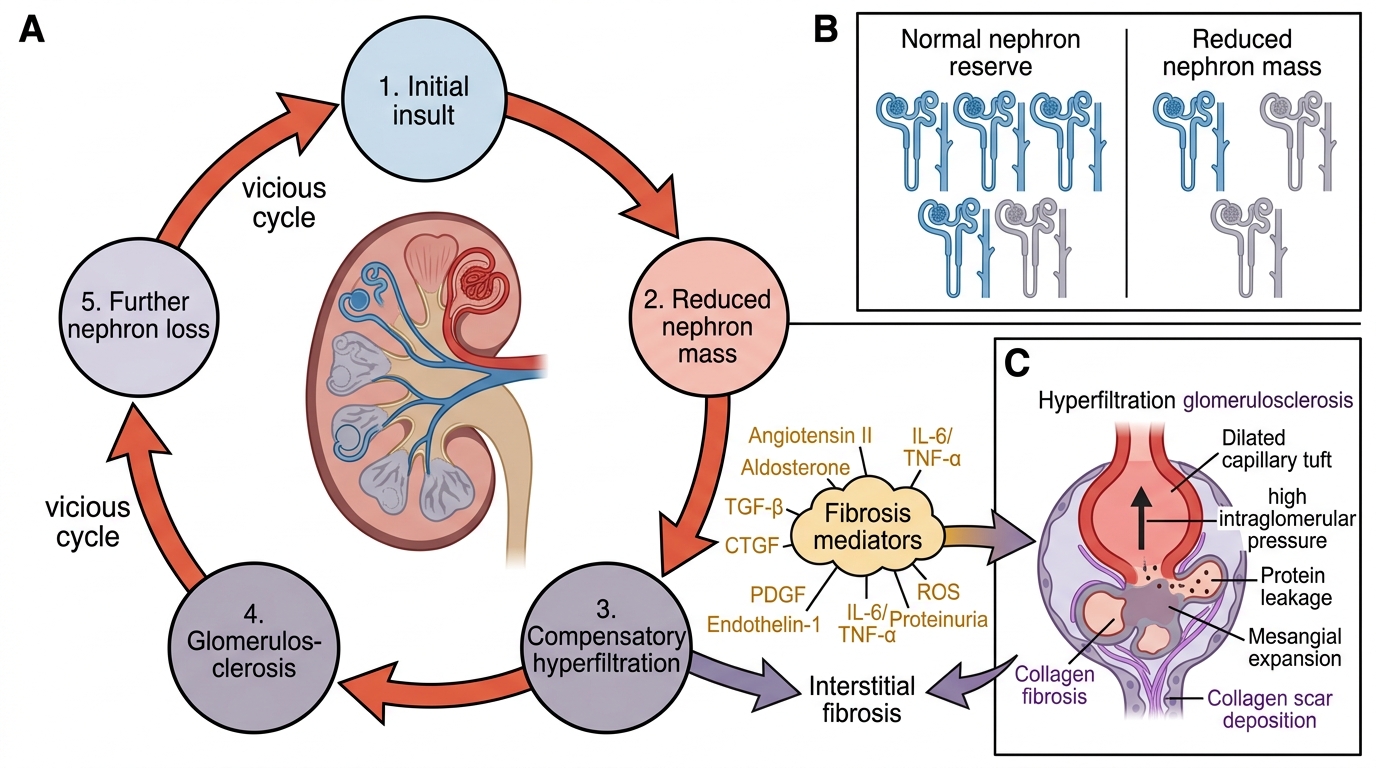
Progressive Nephron Loss in Chronic Kidney Disease
Aetiology — paediatric CKD differs fundamentally from adult CKD:
1. Congenital/structural — CAKUT (Congenital Anomalies of the Kidney and Urinary Tract): the single largest group, accounting for ~50% of paediatric CKD. Includes posterior urethral valves (PUV — the most important single lesion, causing CKD through chronic obstructive nephropathy and pressure damage in utero), vesicoureteric reflux nephropathy (renal scarring from recurrent pyelonephritis), renal dysplasia/hypoplasia, and multicystic dysplastic kidney. Detection often begins antenatally on fetal anomaly ultrasound.
2. Glomerular causes:
• Focal segmental glomerulosclerosis (FSGS): the leading glomerular cause in school-age children; steroid-resistant nephrotic syndrome; progresses to ESRD in >50% over 10 years
• Lupus nephritis: SLE in adolescent girls; immune complex deposits; can rapidly progress to ESRD without aggressive immunosuppression
• Alport syndrome: X-linked (COL4A5 mutation, boys more severely affected); characterised by haematuria + proteinuria + sensorineural hearing loss + anterior lenticonus; no curative therapy; eventual ESRD in 2nd–3rd decade
3. Hereditary/metabolic:
• Autosomal recessive PKD (ARPKD): large echogenic kidneys at birth; liver fibrosis; may present as neonatal respiratory distress from pulmonary hypoplasia
• Cystinosis: lysosomal storage disorder; tubular dysfunction (Fanconi syndrome) early, then glomerular; treated with cysteamine
• Primary hyperoxaluria: oxalate deposits in kidneys; ESRD in childhood; pyridoxine-responsive (type 1)
4. Acquired glomerulonephritis not responding to treatment: HUS (atypical), MPGN, and rapidly progressive GN not in remission.
SELF-CHECK
A 7-year-old boy was diagnosed with posterior urethral valves at birth and underwent valve ablation. He now presents for routine review with creatinine 1.8 mg/dL and eGFR 42 mL/min/1.73m². Which KDIGO stage does he occupy?
A. G2 (eGFR 60–89 mL/min/1.73m²) — mildly decreased
B. G3a (eGFR 45–59 mL/min/1.73m²) — mildly to moderately decreased
C. G3b (eGFR 30–44 mL/min/1.73m²) — moderately to severely decreased
D. G4 (eGFR 15–29 mL/min/1.73m²) — severely decreased
Reveal Answer
Answer: C. G3b (eGFR 30–44 mL/min/1.73m²) — moderately to severely decreased
An eGFR of 42 mL/min/1.73m² falls within the G3b range (30–44 mL/min/1.73m²), which is classified as 'moderately to severely decreased' kidney function. KDIGO stages: G1 ≥90, G2 60–89, G3a 45–59, G3b 30–44, G4 15–29, G5 <15. This boy's CKD from PUV places him at G3b — well beyond the early stages, requiring active management of complications and preparation for possible renal replacement therapy in future years.
Diagnosis and Investigation
Diagnosing CKD in children requires confirming the chronicity of renal dysfunction (ruling out AKI by demonstrating that the impairment has persisted for at least three months), accurately estimating its severity using eGFR staging, identifying the underlying aetiology to guide aetiology-specific therapy, and systematically assessing all major complications so that each can be proactively managed. Each of these objectives requires a distinct but overlapping set of investigations that are performed at diagnosis and repeated at regular intervals.
Confirming and staging CKD — KDIGO eGFR classification:
CKD is defined as kidney damage (structural or functional abnormalities) or eGFR <60 mL/min/1.73m² for ≥3 months. The 3-month duration distinguishes CKD from AKI. In children, eGFR is estimated using the Bedside Schwartz formula: eGFR (mL/min/1.73m²) = 0.413 × height (cm) / serum creatinine (mg/dL). The KDIGO classification uses both GFR stage (G1–G5) and albuminuria category (A1–A3) — the combination determines overall CKD risk and guides monitoring frequency.
| KDIGO GFR Stage | eGFR (mL/min/1.73m²) | Descriptor |
|---|---|---|
| G1 | ≥90 | Normal or high (with kidney damage markers) |
| G2 | 60–89 | Mildly decreased |
| G3a | 45–59 | Mildly to moderately decreased |
| G3b | 30–44 | Moderately to severely decreased |
| G4 | 15–29 | Severely decreased |
| G5 | <15 | Kidney failure (ESRD) |
⚑ AI image — pending faculty review (auto-QA score 7/10; best of 3 attempts)
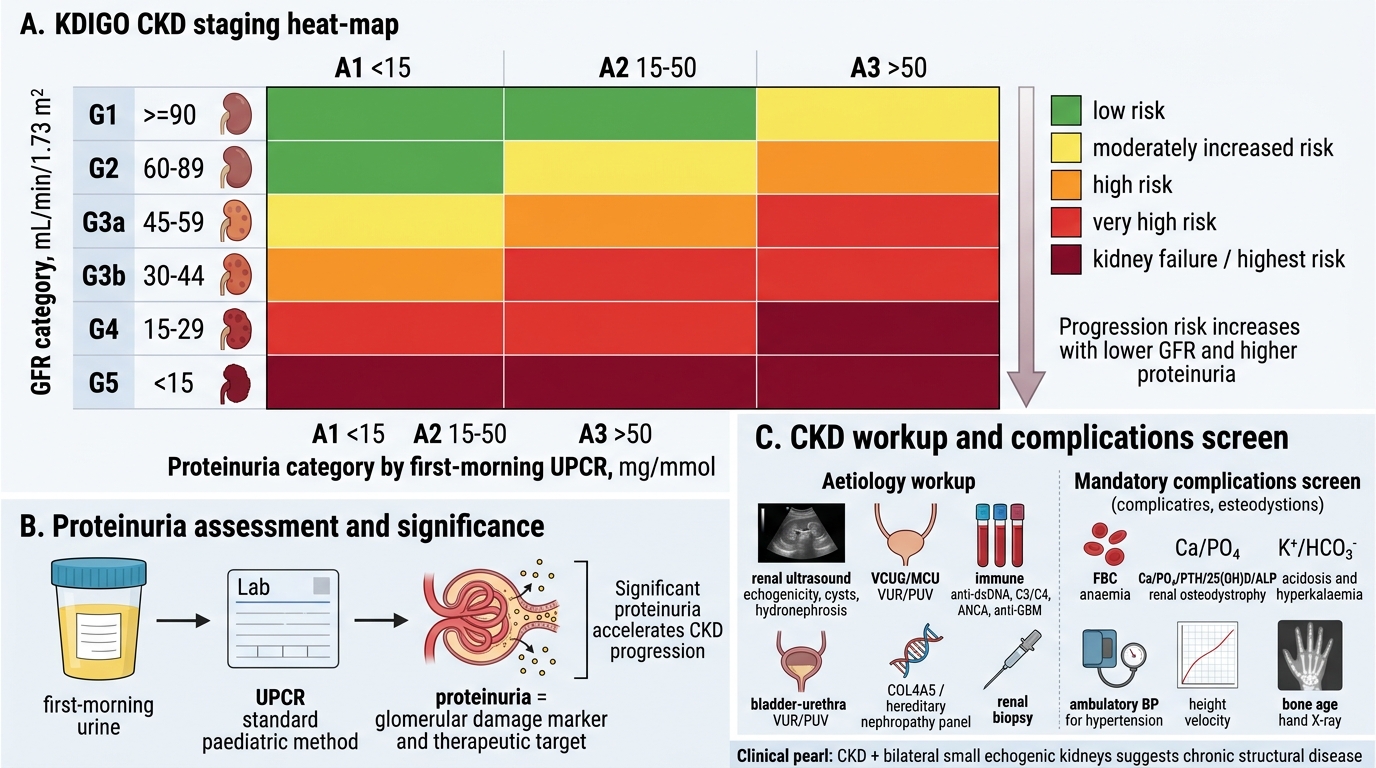
KDIGO CKD Staging and Paediatric Workup
Proteinuria assessment: urine protein:creatinine ratio (UPCR) on a first-morning sample is the standard paediatric method (A1: UPCR <15 mg/mmol; A2: 15–50; A3: >50). Significant proteinuria accelerates CKD progression and is both a marker of glomerular damage and a therapeutic target.
Aetiology workup:
• Renal ultrasonography: size (small = chronic scarring; large = ARPKD/PKD), echogenicity, corticomedullary differentiation, cysts, hydronephrosis
• Voiding cystourethrogram (VCUG/MCU): if VUR or PUV suspected
• Immune profile: ANA, anti-dsDNA, complement C3/C4 (lupus); ANCA (vasculitis); anti-GBM (Goodpasture)
• Genetic studies: COL4A5 sequencing (Alport), genetic panel for hereditary nephropathies
• Renal biopsy: glomerular cause suspected, unexplained CKD, or before starting immunosuppression
Complications workup — mandatory in established CKD:
• FBC: Hb, MCV (normochromic normocytic anaemia in erythropoietin deficiency)
• Calcium, phosphate, PTH, 25(OH)D, alkaline phosphatase (renal osteodystrophy)
• Potassium, bicarbonate (metabolic acidosis, hyperkalaemia)
• 24-hour BP monitoring or ambulatory BP (hypertension)
• Growth chart and height velocity (growth failure)
• Bone age X-ray (delayed skeletal maturation)
CLINICAL PEARL
Small kidneys ≠ always CKD, but CKD + bilateral small echogenic kidneys = structural/chronic disease until proven otherwise. In a child with creatinine elevation, ultrasound finding of bilateral small, hyperechogenic kidneys with loss of corticomedullary differentiation strongly suggests long-standing renal damage — the scarring and fibrosis replace functional parenchyma with fibrous tissue, increasing echogenicity. Conversely, normal-sized or large kidneys with elevated creatinine in a child suggest acute disease (AKI) or infiltrative/cystic disease (PKD, lymphoma). Always correlate kidney size on ultrasound with the clinical timeline before labelling a child's kidney disease as chronic or acute.