Page 19 of 30
PE20.7 | Wilms Tumor — SDL Guide
Learning Objectives
- Describe the clinical presentation of Wilms tumor and identify the key examination trap (do not palpate vigorously)
- Explain the pathophysiology, genetics (WT1), and histology of nephroblastoma including the significance of anaplasia
- Recognise the associated syndromes (WAGR, Beckwith-Wiedemann, Denys-Drash) that increase Wilms tumor risk
- Apply the NWTS staging system (I–V) to determine management: nephrectomy + chemotherapy ± radiation ± nephron-sparing for bilateral disease
- Differentiate Wilms tumor from neuroblastoma on clinical, laboratory, and imaging grounds
INSTRUCTIONS
Wilms tumor — also called nephroblastoma — is the most common primary renal malignancy of childhood and one of the great success stories of paediatric oncology: with multimodal treatment, over 90% of children with localised disease are cured. Yet three traps persist in clinical practice: missing the abdominal mass because it is painless and the child appears well; vigorous abdominal palpation that ruptures the tumour and upstages the disease; and confusing Wilms with neuroblastoma, the two most important causes of abdominal mass in young children. This module gives you the knowledge to avoid all three.
References
- Ghai Essential Pediatrics, 9th ed., Ch. 19 — Paediatric Oncology (textbook)
- Nelson Textbook of Pediatrics, 21st ed., Ch. 522 — Wilms Tumor (textbook)
- Children's Oncology Group (COG) AREN protocols; SIOP Renal Tumour Study Group guidelines (guideline)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A 3-year-old girl is brought by her mother who noticed 'a bump in her tummy' while bathing her. The child appears well, is afebrile, and has been eating normally. On examination there is a large, smooth, non-tender mass in the right flank, clearly staying within the right side of the abdomen and not crossing the midline. Blood pressure is 115/78 mmHg. The intern reaches forward to deeply palpate the mass to better characterise its borders. What should you say to stop him, and why?
WHY THIS MATTERS
Wilms tumor is encountered by every paediatrician because it is common — it accounts for ~6% of all childhood cancers globally and peaks at 3–4 years of age. The dramatic cure rates achieved with modern multimodal therapy (nephrectomy + chemotherapy ± radiation) mean that what the clinician does in the first encounter — specifically, whether the abdomen is palpated gently or vigorously — can directly affect the staging, and therefore the intensity of treatment, that the child will require. Additionally, Wilms tumor has well-characterised genetic and syndromic associations, making it a model for understanding paediatric oncogenetics. Distinguishing it from neuroblastoma — the other major abdominal malignancy of young children — is a core competency tested in examinations and required at the bedside.
RECALL
Before proceeding, recall the following from prior learning:
- Retroperitoneal anatomy: the kidneys are retroperitoneal organs flanking the spine; the adrenal glands sit at their superior poles. Wilms arises from the kidney; neuroblastoma arises from adrenal medulla or paravertebral sympathetic chain.
- Abdominal mass in a toddler: the two most important causes are Wilms tumor (renal, unilateral, smooth, does NOT cross midline) and neuroblastoma (adrenal/paravertebral, may cross midline, irregular, calcification common).
- Tumour suppressor gene concept: WT1 is a tumour suppressor gene on chromosome 11p13; its loss of function contributes to nephroblastoma development through the two-hit hypothesis.
- Paediatric oncology multimodality: surgery + chemotherapy ± radiation is the standard framework; the goal is maximal cure with minimal long-term toxicity.
Clinical Presentation of Wilms Tumor
Wilms tumor (nephroblastoma) is the most common primary renal tumour in children, accounting for approximately 6% of all childhood cancers. It peaks between 3 and 4 years of age, though it can occur from infancy to adolescence. The cardinal and most important presenting feature is an asymptomatic abdominal mass discovered incidentally — most commonly by a parent during bathing, dressing, or play, or by a nurse or physician during a routine physical examination. The fact that the child appears completely well despite a large intraabdominal tumour is a defining and deceptive characteristic: parents often delay seeking attention because the child seems healthy.
The clinical features of Wilms tumor span renal, vascular, and constitutional domains, and can be organised as follows:
- Abdominal mass: large, smooth, firm, and well-defined; confined to one side (unilateral in ~94% of cases); does NOT cross the midline (contrast with neuroblastoma); non-tender; moves with respiration
- Haematuria: microscopic (~25%) or macroscopic (~12%); may be the presenting symptom without a palpable mass
- Hypertension: in ~25% of cases, due to renin secretion by the tumour or compression of the renal artery; may cause headache
- Abdominal pain: uncommon; if acute pain occurs, consider spontaneous intratumoral haemorrhage or rupture
- Fever and weight loss: suggest advanced or bilateral disease
- Constitutional symptoms (malaise, anorexia) are unusual and indicate extensive disease
CRITICAL EXAMINATION RULE: Do NOT vigorously palpate the abdomen of a child in whom Wilms tumor is suspected. Vigorous palpation risks tumour capsule rupture, which releases malignant cells into the peritoneum, converts the tumour from Stage I/II to Stage III, and significantly increases the intensity (and toxicity) of required treatment. Examination should be gentle and limited to confirming the presence and side of the mass.
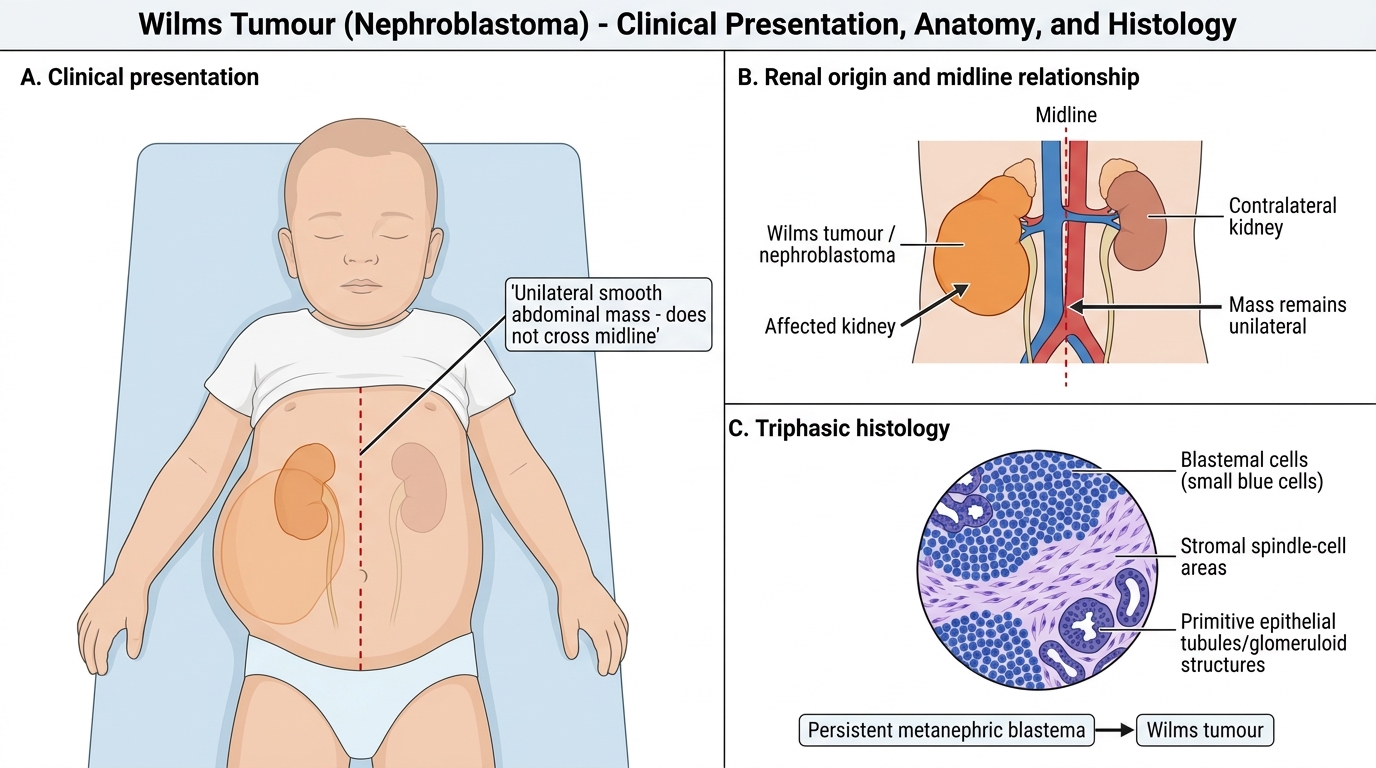
Wilms Tumour: Unilateral Abdominal Mass and Triphasic Histology
Pathophysiology, Genetics, and Associated Syndromes
Wilms tumor is a mixed embryonal tumour that arises from persistent metanephric blastema — the embryonic precursor cells of the kidney — that fail to differentiate normally during nephrogenesis. This explains both its occurrence in young children and its characteristic histological composition.
Histology — triphasic pattern:
The classic Wilms tumour is composed of three elements in varying proportions, reflecting the three lineages of normal kidney development. This triphasic pattern is diagnostically characteristic and distinguishes nephroblastoma from other paediatric renal masses — a purely monophasic or biphasic tumour should prompt consideration of clear cell sarcoma or rhabdoid tumour, which have different treatment protocols. The three distinct components, described here in order of their prognostic and biological significance in Wilms tumor research and treatment stratification, are:
• Blastemal component: small, round, primitive blue cells — the undifferentiated metanephric blastema; drives tumour growth and chemosensitivity
• Stromal component: spindle cells, smooth muscle, adipose tissue — mesenchymal derivatives
• Epithelial component: forming primitive tubules and glomeruli — partially differentiated towards renal epithelium
Anaplasia (nuclear enlargement ≥3× normal + hyperchromatic multipolar mitoses) is the key adverse histological feature and is found in ~5–10% of cases. Diffuse anaplasia carries a significantly worse prognosis and requires more intensive therapy. Focal anaplasia is intermediate-risk. Absence of anaplasia = favourable histology with excellent cure rates.
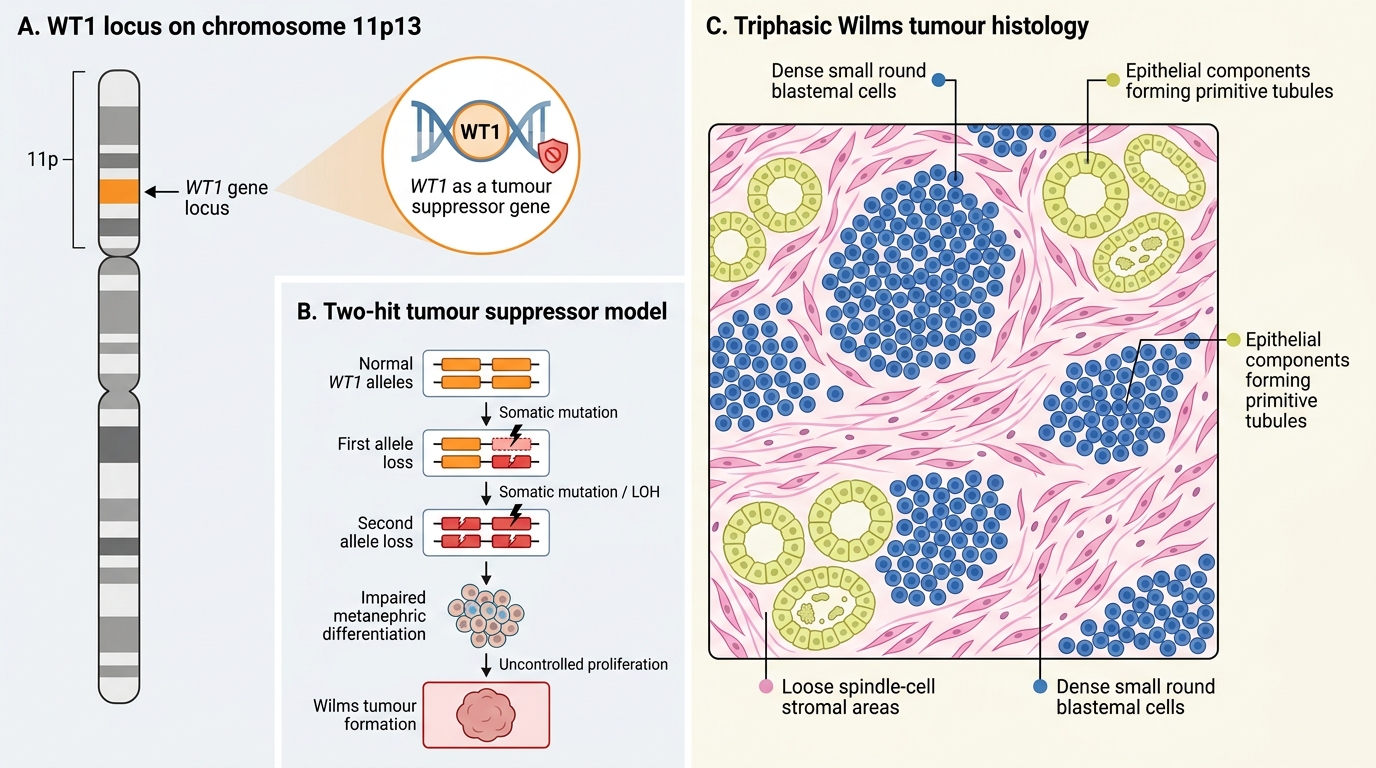
WT1 Genetics and Triphasic Wilms Tumour Histology
Genetics:
WT1 (Wilms Tumor gene 1) on chromosome 11p13 is the key tumour suppressor gene. Loss of both alleles (two-hit model) disrupts normal metanephric differentiation and drives tumorigenesis. However, WT1 is mutated in only ~10–15% of sporadic Wilms tumours; other loci (WT2 at 11p15, WTX, CTNNB1) are involved in the majority. The sporadic form is the most common (~95%); familial Wilms is rare.
Associated syndromes:
| Syndrome | Features | Wilms Risk |
|---|---|---|
| WAGR syndrome | Wilms + Aniridia + Genito-urinary anomalies + intellectual Retardation; WT1 deletion at 11p13 | ~50% |
| Beckwith-Wiedemann syndrome (BWS) | Macroglossia, hemihypertrophy, omphalocele, hyperinsulinaemia (neonatal hypoglycaemia); WT2 locus (IGF2 upregulation) | ~5–10% |
| Denys-Drash syndrome | Diffuse mesangial sclerosis (nephrotic syndrome) + pseudohermaphroditism + Wilms; WT1 point mutation | >90% |
Children with these syndromes require surveillance ultrasonography every 3 months until age 7–8 years. Bilateral Wilms tumor (Stage V, ~6% of cases) is more common in syndromic patients.
SELF-CHECK
A 2-year-old boy has bilateral abdominal masses on ultrasound suggestive of bilateral Wilms tumor. He has macroglossia, hemihypertrophy of his left side, and a history of neonatal hypoglycaemia. Which syndrome does he have?
A. WAGR syndrome (Wilms, Aniridia, GU anomalies, intellectual Retardation)
B. Beckwith-Wiedemann syndrome (macroglossia, hemihypertrophy, omphalocele, hyperinsulinaemia)
C. Denys-Drash syndrome (nephrotic syndrome, pseudohermaphroditism, Wilms)
D. Li-Fraumeni syndrome (TP53 germline mutation, multiple malignancy predisposition)
Reveal Answer
Answer: B. Beckwith-Wiedemann syndrome (macroglossia, hemihypertrophy, omphalocele, hyperinsulinaemia)
The triad of macroglossia + hemihypertrophy + neonatal hypoglycaemia (from hyperinsulinaemia due to nesidioblastosis) is classic Beckwith-Wiedemann syndrome (BWS). BWS results from IGF2 upregulation at the WT2 locus (11p15) and carries a 5–10% risk of Wilms tumor (bilateral cases are more common in BWS than in sporadic Wilms). WAGR requires aniridia and GU anomalies. Denys-Drash presents with nephrotic syndrome and intersex. Li-Fraumeni is a cancer predisposition syndrome but not specifically associated with these features.
Diagnosis, Investigation, and Staging
Diagnosis of Wilms tumor proceeds through a structured sequence of imaging and staging investigations. Importantly, in most centres following COG (American) protocols, nephrectomy is performed upfront without pre-operative biopsy, with pathological staging confirmed at surgery; in centres following SIOP (European) protocols, pre-operative chemotherapy is given to shrink the tumour before surgery. Understanding both approaches is required for examinations.
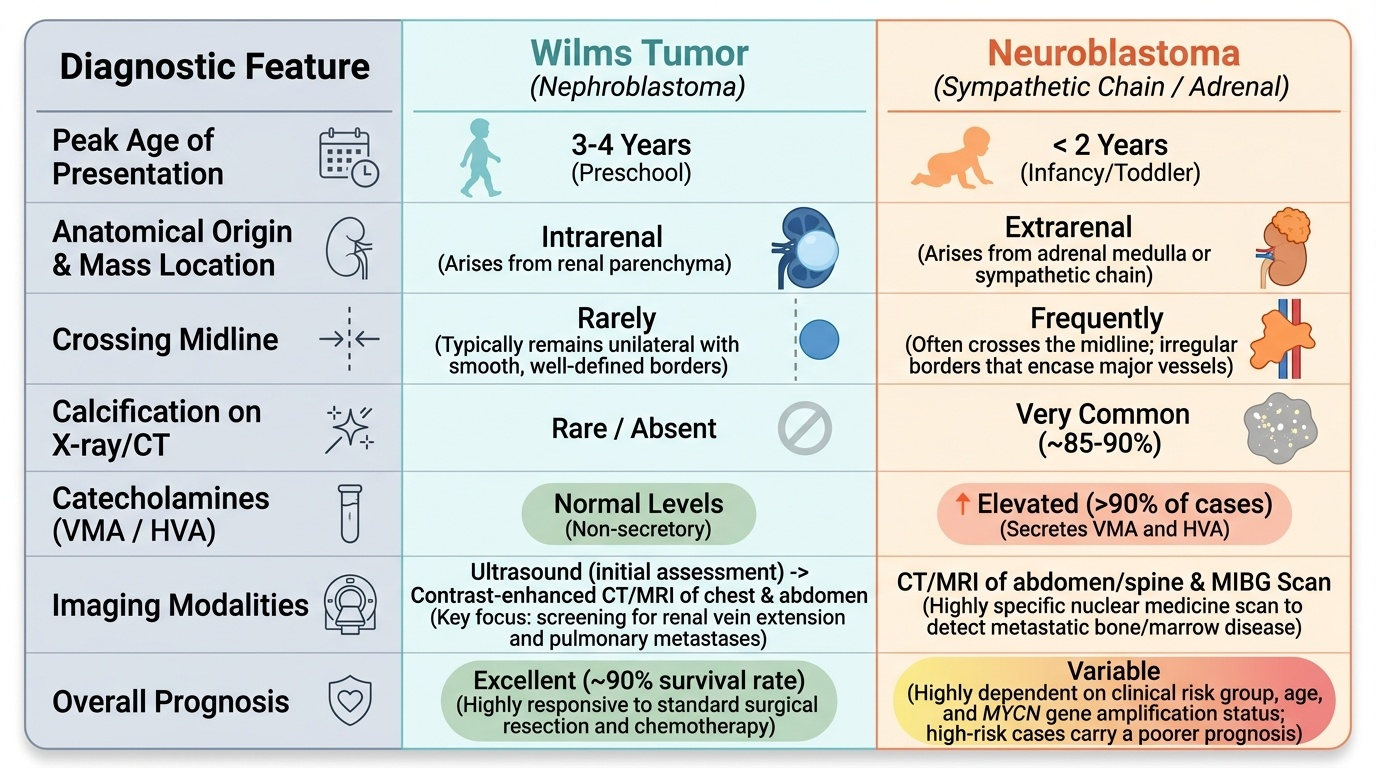
Provided image
Imaging workup:
Imaging in Wilms tumor serves two sequential purposes: first, to characterise the primary renal mass and confirm its intrarenal origin; second, to stage the disease by identifying local extension, lymph node involvement, vascular invasion, and distant metastases. Ultrasound is always the first modality, with CT providing definitive staging. The key imaging findings that support Wilms over neuroblastoma on ultrasound are: intrarenal origin, smooth well-defined borders, and absence of calcification.
• Abdominal ultrasound (first-line imaging): confirms a solid intrarenal mass, assesses the contralateral kidney (bilateral disease?), tumour vascularity (Doppler), renal vein or IVC extension (tumour thrombus is present in ~4% of cases)
• Contrast-enhanced CT of chest and abdomen: defines the extent of local disease (capsular breach, lymph node involvement, adjacent organ invasion), detects pulmonary metastases (the most common site of haematogenous spread)
• Chest X-ray: pulmonary metastases present as round nodules; mandatory initial staging
• Bone scan and brain imaging only if clinically indicated (bone mets rare)
NWTS Staging (National Wilms Tumor Study / Children's Oncology Group):
| Stage | Definition |
|---|---|
| I | Tumour confined to kidney and completely resected; no capsule breach; no vascular involvement |
| II | Extends beyond renal capsule but completely resected (local spread but negative margins) |
| III | Residual non-haematogenous tumour in abdomen (peritoneal spill/biopsy, positive lymph nodes, incomplete resection) |
| IV | Haematogenous metastases (lung, liver, bone, brain) |
| V | Bilateral Wilms tumor |
Differentiating Wilms tumor from neuroblastoma — the two most important abdominal masses in young children:
Key distinguishing features at a glance:
• Wilms: smooth mass stays within one flank, does NOT cross midline; no calcification (rare); urinary catecholamines NORMAL; arises from kidney (intrinsic); better prognosis
• Neuroblastoma: irregular, may cross midline, CALCIFICATION on plain X-ray (~85%), urinary VMA/HVA (homovanillic + vanillylmandelic acid) ELEVATED; arises from adrenal or sympathetic chain; worse prognosis in advanced disease
CLINICAL PEARL
Crossing the midline is the neuroblastoma clue. When a child presents with an abdominal mass, the single most important clinical question at the bedside is: does the mass cross the midline? A smooth, non-tender mass confined to one flank that does NOT cross midline strongly suggests Wilms tumor (renal origin, well-encapsulated). A mass that does cross the midline — especially if irregular, accompanied by skin nodules (blueberry muffin baby), periorbital ecchymosis (panda eyes), or hypertension with sweating — should prompt immediate evaluation for neuroblastoma, including urine catecholamines (VMA/HVA). This bedside distinction guides urgent imaging and prevents diagnostic delay in neuroblastoma, which has a very different treatment pathway.