Page 20 of 42
PE23.12 | Malabsorption — SDL Guide
Learning Objectives
- Describe the clinical features of malabsorption syndrome in children and identify the systemic effects of chronic fat, carbohydrate, and micronutrient malabsorption
- Explain the pathogenesis of coeliac disease (gluten-sensitive enteropathy) and the Marsh classification of duodenal biopsy changes
- Outline the pathogenesis of malabsorption in cystic fibrosis and distinguish it from coeliac disease
- Interpret the diagnostic investigations for coeliac disease (anti-tTG IgA with total IgA, duodenal biopsy) and cystic fibrosis (sweat chloride >60 mmol/L)
- Describe the management principles: gluten-free diet for coeliac disease and pancreatic enzyme replacement for cystic fibrosis
INSTRUCTIONS
Malabsorption in childhood presents as a diagnostic challenge: the child is not absorbing the nutrients essential for growth, yet the symptoms — loose stools, poor weight gain, abdominal distension — overlap with common infections and functional GI disorders. The two most important treatable causes in Indian paediatric practice are coeliac disease (gluten-sensitive enteropathy) and cystic fibrosis, both of which require specific investigations and lifelong management. Recognising the clinical pattern early and initiating the correct diagnostic pathway prevents the irreversible consequences of prolonged nutritional deficiency in a growing child.
References
- Ghai Essential Pediatrics, 9th Ed, Ch 13 (textbook)
- Nelson Textbook of Pediatrics, 21st Ed, Ch 371–375 (textbook)
- ESPGHAN Guidelines on Diagnosis of Coeliac Disease in Children, 2020 (guideline)
- IAP Guidelines on Cystic Fibrosis Management in Children (guideline)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A 3-year-old girl is referred to the paediatric outpatient clinic because she has 'never been well since she started solid foods at 6 months.' Her mother reports she has always had frequent, pale, bulky, foul-smelling stools that float in the toilet. She is markedly thin, her abdomen is protuberant, and her buttocks appear wasted. Her weight is at the 2nd centile (where she had been on the 50th centile at birth), and her height is significantly below the 3rd centile. She is irritable and has two to three soft, bulky stools daily. The family eats wheat-based foods (roti, bread) as their primary staple. Her haemoglobin is 9 g/dL and MCV is elevated. A serum calcium is borderline low. What is the unifying diagnosis and what single blood test would you order first to confirm it?
WHY THIS MATTERS
Malabsorption in childhood is not a rare disease encountered only at tertiary centres — coeliac disease affects approximately 1% of the global population and is significantly under-diagnosed in India because its presentation in Indian children differs from the classic Western phenotype (overt steatorrhoea is less common; growth failure and anaemia often predominate). Cystic fibrosis, though less common in India, carries a high morbidity burden and is frequently misdiagnosed as recurrent respiratory infections without a malabsorptive component being recognised. Both conditions are lifelong, both are treatable, and both carry serious consequences if the diagnosis is delayed — coeliac disease is associated with lymphoma risk if untreated into adulthood, and cystic fibrosis with progressive bronchiectasis and pancreatic destruction. The key clinical skill is recognising the malabsorption syndrome pattern (failure to thrive + abnormal stools + micronutrient deficiency signs) and selecting the correct first-line investigation.
RECALL
Before engaging with this module, recall the normal structure of the small intestinal mucosa: villi project into the intestinal lumen and are lined by enterocytes that express brush border enzymes (lactase, sucrase, maltase) and absorb nutrients; the villi provide an enormous absorptive surface area. Fat-soluble vitamins A, D, E, and K require intact fat absorption and bile salts — any cause of fat malabsorption will lead to fat-soluble vitamin deficiencies. Vitamin B12 is absorbed specifically at the terminal ileum via intrinsic factor; folate is absorbed in the upper small intestine. Iron is absorbed in the duodenum and proximal jejunum. Recall the signs of specific nutrient deficiencies: Vitamin D deficiency (rickets — bowing of legs, Harrison's sulcus); Vitamin K deficiency (bleeding tendency, raised PT); Vitamin A deficiency (night blindness, Bitot's spots); iron deficiency (hypochromic microcytic anaemia); B12/folate deficiency (macrocytic megaloblastic anaemia). Recall that malnutrition (SAM) is defined by MUAC <11.5 cm and/or weight-for-height < −3 SD — chronic malabsorption is a secondary cause of SAM in children.
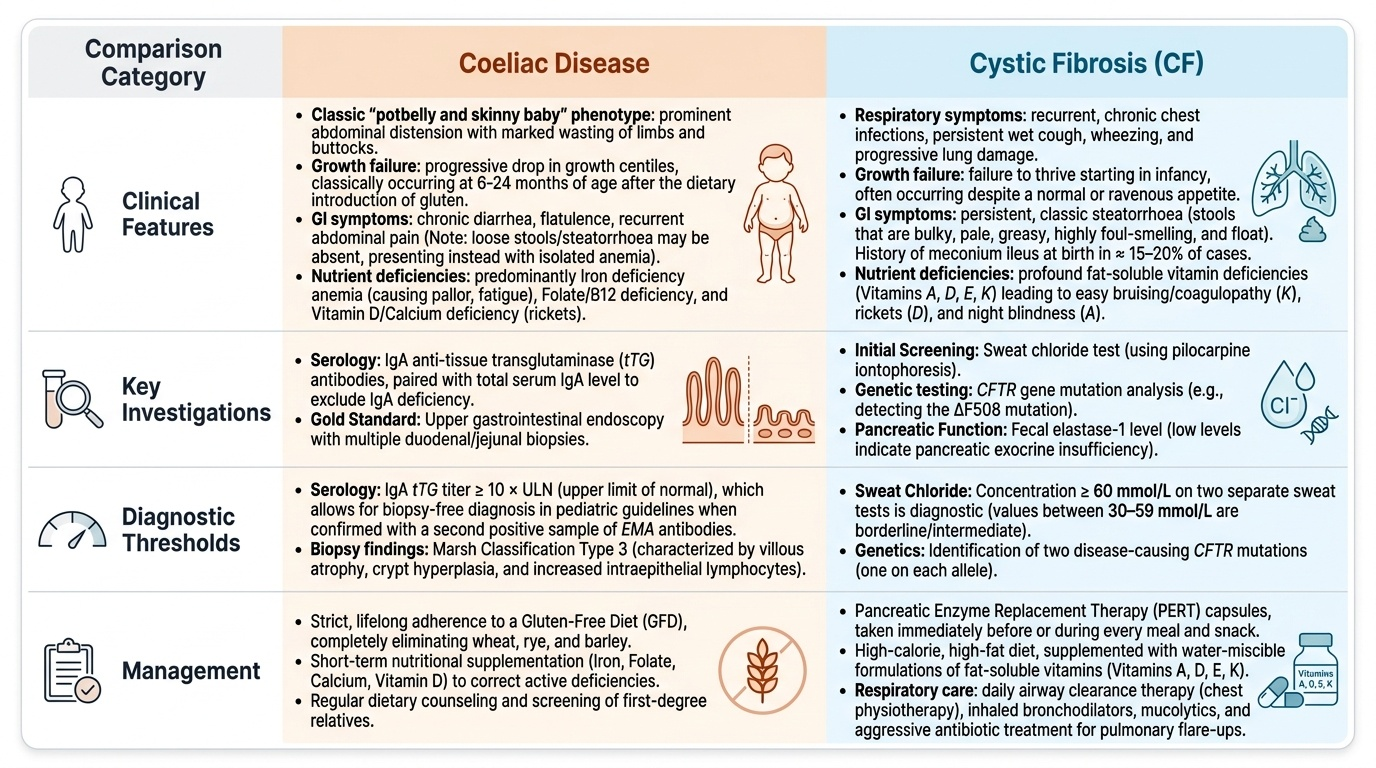
Clinical Presentation of Malabsorption
The clinical hallmark of malabsorption in childhood is a constellation of signs reflecting impaired nutrient absorption across multiple categories: macronutrients (fat, carbohydrates, protein), micronutrients (vitamins, minerals), and their downstream effects on growth and end-organ function. The presentation varies with age at onset, the underlying cause, and the duration of untreated disease, making a systematic clinical assessment essential.
Provided image
Growth failure is the most consistent and often the earliest sign. A child who was growing on a normal centile and then drops progressively — especially after a dietary change (introduction of gluten-containing foods in coeliac disease; introduction of formula in CF-associated malabsorption) — should prompt consideration of malabsorption. Weight loss or failure to gain weight proceeds height retardation in most cases; the classic picture is a thin child with a disproportionately large protruberant abdomen (from gaseous distension and loss of abdominal muscle bulk).
Steatorrhoea — loose, pale, bulky, greasy, foul-smelling stools that may float — is the characteristic stool of fat malabsorption. Not all children with malabsorption have obvious steatorrhoea; in coeliac disease in India, loose stools may be absent and the presentation may be primarily anaemia and growth failure. However, when steatorrhoea is present, it is highly specific.
Signs of specific deficiency states reflect the particular nutrients being malabsorbed:
- Fat-soluble vitamins: Rickets (Vitamin D), coagulopathy/bruising (Vitamin K), night blindness/Bitot's spots (Vitamin A), peripheral neuropathy (Vitamin E).
- Iron deficiency anaemia: pallor, fatigue, irritability, tachycardia (most common in coeliac disease).
- Folate/B12 deficiency: macrocytic anaemia, glossitis.
- Hypoalbuminaemia (protein malnutrition): oedema, ascites in severe cases.
- Calcium/Vitamin D deficiency: hypocalcaemia, tetany, rickets.
Additional features that point to specific diagnoses: abdominal distension with wasted limbs and buttocks ('potbelly + skinny baby' phenotype) is classic for coeliac disease in young children. Recurrent chest infections in association with malabsorption should trigger cystic fibrosis screening. Family history of coeliac disease (first-degree relative) significantly increases pretest probability.
| Clinical feature | Mechanism | Points to |
|---|---|---|
| Pale, floating, bulky stools | Steatorrhoea (fat malabsorption) | CF, coeliac, bile acid deficiency |
| Growth failure after gluten introduction | Villous atrophy impairing absorption | Coeliac disease |
| Recurrent chest infections + steatorrhoea | Pancreatic exocrine insufficiency + bronchiectasis | Cystic fibrosis |
| Iron deficiency anaemia without obvious blood loss | Duodenal/jejunal absorptive failure | Coeliac disease |
| Oedema in a thin child | Hypoalbuminaemia from protein malabsorption | Severe/prolonged malabsorption |
Pathogenesis and Aetiology
Understanding the pathogenic mechanism of malabsorption in each condition is essential for rational test selection and interpreting laboratory results. The two dominant conditions — coeliac disease and cystic fibrosis — have entirely different mechanisms that produce the same end result of malabsorption.
Coeliac disease is an immune-mediated, gluten-sensitive enteropathy. Gliadin, a storage protein of wheat (and related prolamins from rye and barley), is only partially digested in the small intestine. In genetically predisposed individuals (HLA-DQ2 and DQ8 haplotypes account for >90% of patients), these gliadin peptides are deamidated by the enzyme tissue transglutaminase (tTG) in the intestinal lamina propria. The deamidated peptides are presented to CD4+ T lymphocytes via HLA-DQ2/DQ8, triggering an adaptive immune response. This response damages the enterocytes directly and stimulates CD8+ cytotoxic T cells, leading to: (1) increased intraepithelial lymphocytes (the earliest change); (2) crypt hyperplasia; and (3) progressive villous atrophy — the hallmark of established coeliac disease. Villous atrophy reduces the absorptive surface area of the small intestine by up to 90%, causing pan-malabsorption. The immune response also generates antibodies: IgA anti-tTG antibodies (the serum marker) and IgA anti-endomysial antibodies (EMA). Autoimmune targeting also explains coeliac disease's association with other autoimmune conditions (Type 1 diabetes, thyroid disease).
Cystic fibrosis (CF) is a multisystem autosomal recessive disease caused by mutations in the CFTR gene, which encodes a chloride channel in epithelial cells. Loss of CFTR function causes abnormally thick, viscous mucus across multiple organs. In the pancreas, thick mucus obstructs pancreatic ducts, leading to progressive pancreatic exocrine insufficiency — the pancreas cannot secrete adequate lipase, protease, and amylase into the duodenum. Without digestive enzymes, fat and protein are not broken down and cannot be absorbed. Simultaneously, CF causes abnormally viscous airway secretions, recurrent pulmonary infections, and progressive bronchiectasis — explaining the combined malabsorptive and respiratory phenotype.
Post-infectious enteropathy (tropical enteropathy/persistent diarrhoea) is a clinically important cause in India: acute gastroenteritis (viral, bacterial, or parasitic) damages the small intestinal villous architecture. If this damage is prolonged or repeated, persistent malabsorption ensues — particularly lactose malabsorption from secondary lactase deficiency and secondary fat malabsorption. This is common in malnourished children in low-resource settings.
Other causes include small intestinal bacterial overgrowth (SIBO — bacteria ferment sugars before absorption, causing bloating and fat malabsorption), abetalipoproteinaemia (rare, cannot form chylomicrons — fat is trapped in enterocytes and cannot exit into the lymphatics), and Schwachman-Diamond syndrome (pancreatic exocrine insufficiency with bone marrow failure).
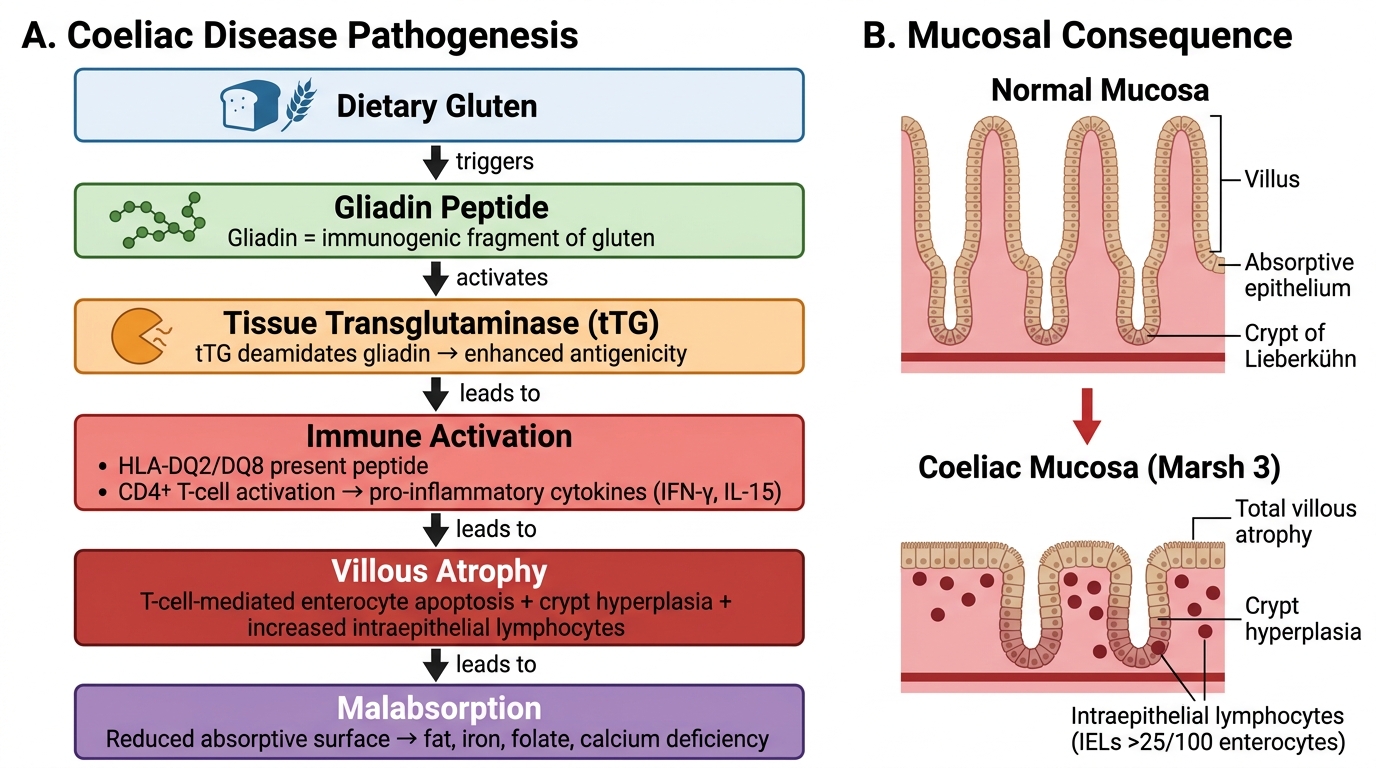
Coeliac Disease: Pathogenesis Cascade and Mucosal Consequence
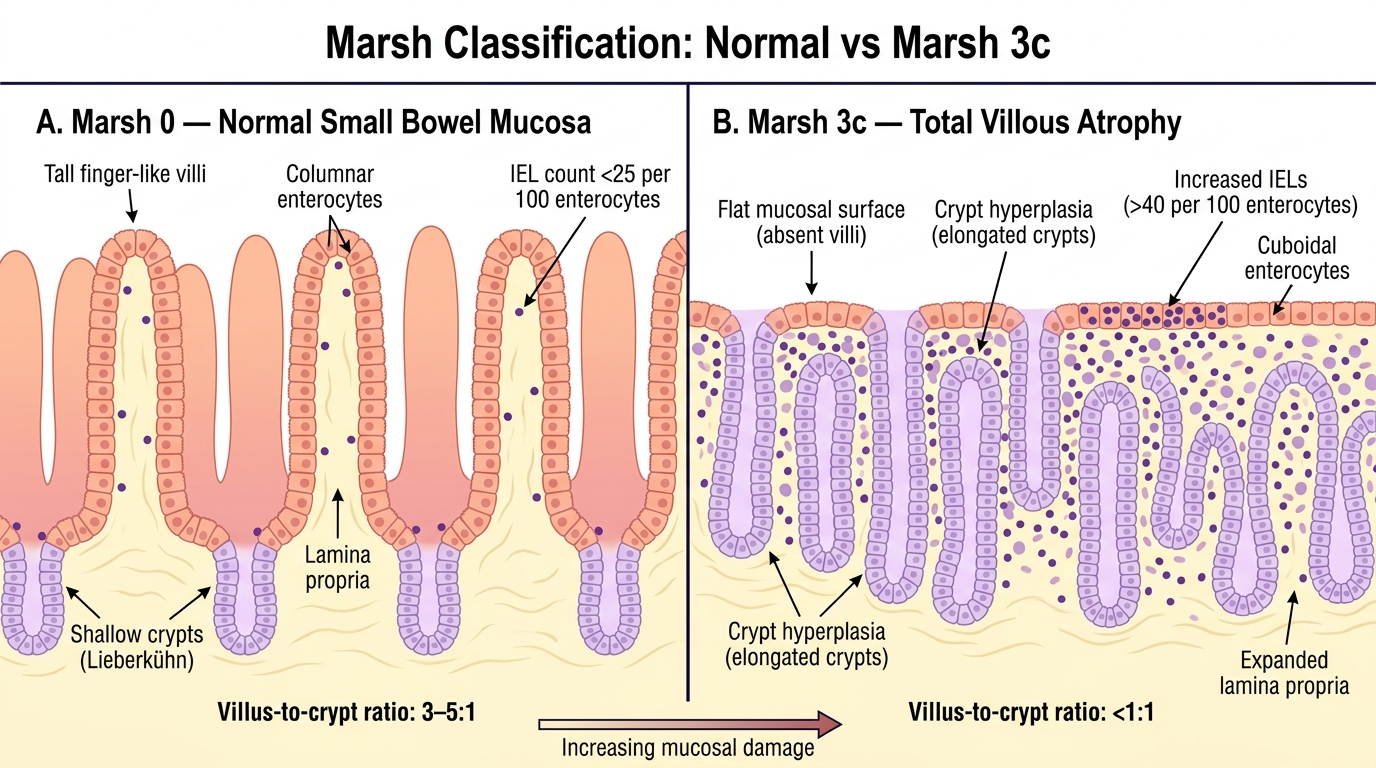
Marsh Classification: Normal Small Bowel Mucosa (Marsh 0) vs Total Villous Atrophy (Marsh 3c)
SELF-CHECK
A 4-year-old girl has been diagnosed with coeliac disease. Her serum anti-tTG IgA was markedly elevated. Her duodenal biopsy shows flattened villi, crypt hyperplasia, and >40 intraepithelial lymphocytes per 100 enterocytes. Which Marsh grade does this describe?
A. Marsh 1 — increased intraepithelial lymphocytes only
B. Marsh 2 — crypt hyperplasia with normal villi
C. Marsh 3c — total villous atrophy with crypt hyperplasia and increased IELs
D. Marsh 4 — hypoplastic atrophy
Reveal Answer
Answer: C. Marsh 3c — total villous atrophy with crypt hyperplasia and increased IELs
Marsh 3 describes villous atrophy, which is subclassified as 3a (partial), 3b (subtotal), and 3c (total villous atrophy). The description — flattened villi + crypt hyperplasia + increased intraepithelial lymphocytes — matches Marsh 3 (total villous atrophy = Marsh 3c). Marsh 1 has increased IELs but normal villi and no crypt hyperplasia. Marsh 2 has crypt hyperplasia but the villi are preserved. Marsh 4 (hypoplastic atrophy — rare, severe) is not the typical active coeliac presentation.
Diagnosis and Investigation
Accurate diagnosis of the cause of malabsorption requires a stepwise investigative approach: begin with non-invasive tests that have high sensitivity and specificity, then proceed to confirmatory testing. A rational investigation sequence prevents unnecessary invasive procedures while ensuring that treatable causes are identified promptly and that the burden on the child and family is minimised. The investigations serve a dual purpose: first, confirming that malabsorption is genuinely present (as opposed to constitutional small stature or other causes of poor growth); second, identifying the specific pathogenic mechanism. Each cause of malabsorption has a characteristic pattern of findings, and correlating the clinical history (dietary gluten exposure, recurrent chest infections, family history) with the laboratory results allows a confident diagnosis in the majority of cases without delay and before invasive biopsy is required.
Confirming malabsorption:
- Stool Sudan stain for fat (microscopy): presence of fat globules and fatty acid crystals confirms steatorrhoea. A positive Sudan stain is highly suggestive of fat malabsorption.
- D-xylose absorption test: D-xylose is absorbed passively in the proximal small intestine without enzymatic digestion. Low urinary D-xylose excretion (<15 g in 5 hours) after a 25 g oral dose indicates mucosal disease (coeliac, post-infectious enteropathy) rather than pancreatic exocrine insufficiency.
- Stool for reducing substances: positive reducing substances (Clinitest) indicate carbohydrate malabsorption (lactose malabsorption). Useful in post-infectious enteropathy.
Coeliac disease:
- Serology (first-line): anti-tTG IgA is the recommended screening test — high sensitivity (>95%) and specificity. CRITICAL: total serum IgA must be measured simultaneously because IgA-deficient patients (estimated 2–3% of coeliac patients) will have a false-negative anti-tTG IgA. If total IgA is low (<7 mg/dL), test anti-tTG IgG or anti-deamidated gliadin peptide (anti-DGP) IgG instead.
- Anti-endomysial antibodies (EMA IgA): highly specific (near 100%) but less sensitive; used as a second-line confirmatory test.
- Duodenal biopsy (confirmatory): endoscopic biopsy of the second/third part of the duodenum remains the gold standard. Multiple specimens (≥4) from different sites are taken because of patchy involvement. Histology is classified by the Marsh classification (0–4). Coeliac disease is established by Marsh 2–4 in the context of positive serology. ESPGHAN 2020 guidelines allow coeliac diagnosis without biopsy if anti-tTG IgA is >10× upper limit of normal (ULN) AND EMA IgA is positive AND the child is symptomatic — this applies to paediatric GI specialist evaluation.
- HLA typing: HLA-DQ2/DQ8 testing has very high negative predictive value — absence of DQ2/DQ8 makes coeliac disease very unlikely; but it cannot confirm the diagnosis (many people carry DQ2/DQ8 without coeliac disease).
Cystic fibrosis:
- Sweat chloride test (gold standard): pilocarpine iontophoresis collects sweat, and chloride concentration is measured. >60 mmol/L is diagnostic; 30–59 mmol/L is borderline (repeated testing needed); <30 mmol/L is normal. The test must be performed with adequate sweat volume (>75 mg); invalid if volume insufficient.
- CFTR mutation analysis: identifies the specific CFTR mutation (>2,000 known); delta-F508 is the most common mutation globally. Important for prognosis and emerging targeted therapies (CFTR modulators like ivacaftor, lumacaftor).
- Newborn screening (IRT): immunoreactive trypsinogen on dried blood spot (Guthrie card) — elevated IRT prompts confirmation by sweat chloride test and CFTR genetics.
- Faecal elastase-1: low (<100 µg/g stool) confirms pancreatic exocrine insufficiency; supports CF diagnosis in context.
General nutritional assessment: Weight-for-height Z-score, MUAC, serum albumin, CBC (anaemia pattern), serum calcium, Vitamin D (25-OH), Vitamin A, PT/INR (Vitamin K status), serum zinc.
CLINICAL PEARL
Always measure total serum IgA alongside anti-tTG IgA — this is the most commonly missed step in coeliac screening. Selective IgA deficiency occurs in 2–3% of coeliac disease patients (up to 10 times the prevalence in the general population), and these patients will have a normal or undetectable anti-tTG IgA despite active coeliac disease. If you order anti-tTG IgA alone and the patient is IgA-deficient, you will have a falsely reassuring normal result. The solution is simple: always add total serum IgA to the request. If total IgA is deficient, switch to IgG-based testing (anti-tTG IgG or anti-DGP IgG). A second key trap: children on a gluten-free diet before serology is performed will have negative anti-tTG IgA — the antibody titres fall rapidly after gluten withdrawal. Always perform serology WHILE the patient is eating gluten (a gluten challenge may be needed if the diet has already been changed).