Page 12 of 31
PE24.4 | Status Epilepticus — SDL Guide
Learning Objectives
- Define status epilepticus using the operational (≥5 minutes) and physiological (>30 minutes) definitions
- Describe the aetio-pathogenesis and clinical approach to status epilepticus in children
- Apply the time-based management algorithm for status epilepticus, including drug selection, weight-based dosing, and routes of administration
- Identify the metabolic causes of seizures that must be corrected concurrently with antiepileptic drug therapy
- Recognise refractory status epilepticus and describe the indications for escalating to intensive care
INSTRUCTIONS
Status epilepticus is one of the most common true paediatric neurological emergencies and a frequent MBBS examination scenario. The operational definition was revised in 2015 by the ILAE to ≥5 minutes — earlier than the traditional 30 minutes — because seizures that last more than 5 minutes rarely stop spontaneously and become progressively harder to treat as time passes. A time-based, drug-stepwise algorithm governs management; memorising the drug sequence and doses is a core clinical competency for every doctor who may encounter a seizing child.
References
- Ghai Essential Pediatrics, 9th ed., Ch. 14 — Seizure Disorders (textbook)
- Nelson Textbook of Pediatrics, 21st ed., Ch. 621 — Status Epilepticus (textbook)
- ILAE 2015 Definition of Status Epilepticus — Trinka et al., Epilepsia 2015 (guideline)
- ConSEPT/EcLiPSE Trials 2019 — Levetiracetam vs Phenytoin in Paediatric SE (guideline)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A 4-year-old child with a history of febrile convulsions is brought to the emergency department by ambulance. The paramedics report the child has been having a generalised tonic-clonic seizure for 8 minutes continuously without stopping. He is cyanosed, with oxygen saturation of 83%. He is still convulsing on arrival. The nurse asks you what drug to give. You have IV access. What is your immediate management?
WHY THIS MATTERS
Status epilepticus (SE) is the most common life-threatening neurological emergency in children, with an incidence of approximately 10–40 per 100,000 children per year. It is associated with significant morbidity — hypoxic-ischaemic brain injury, aspiration pneumonia, hyperthermia, and metabolic derangements — and mortality of approximately 3–5% in developed countries (higher in resource-limited settings). The risk of permanent neurological sequelae rises with duration of SE; each minute of uncontrolled seizure worsens the physiological milieu. For a final-year student, knowing the time-based algorithm, the correct first-line drug and dose by access route, and the metabolic causes that must be corrected simultaneously is essential for safe management in any paediatric posting.
RECALL
Before proceeding, recall the following:
• Seizure mechanism: abnormal, excessive, or synchronous neuronal discharge; GABA (inhibitory) and glutamate (excitatory) are the primary neurotransmitters; benzodiazepines potentiate GABA-A receptor function by increasing chloride channel opening frequency — this is the mechanism of action of the first-line drugs in SE.
• Seizure classification: focal (involves one hemisphere, may or may not involve loss of awareness) vs generalised (both hemispheres from onset — includes tonic-clonic, tonic, clonic, absence, atonic, myoclonic).
• Febrile seizures: the commonest seizure type in children 6 months–5 years; simple febrile seizure (<15 min, generalised, single per febrile illness); complex febrile seizure (>15 min, or focal, or >1 in 24 h).
• Metabolic causes of seizures: hypoglycaemia (most urgent — treat before anything else), hyponatraemia (Na <125 mmol/L), hypocalcaemia (Ca <1.8 mmol/L), hypomagnesaemia, pyridoxine deficiency (neonates).
Clinical Presentation: Recognising Status Epilepticus
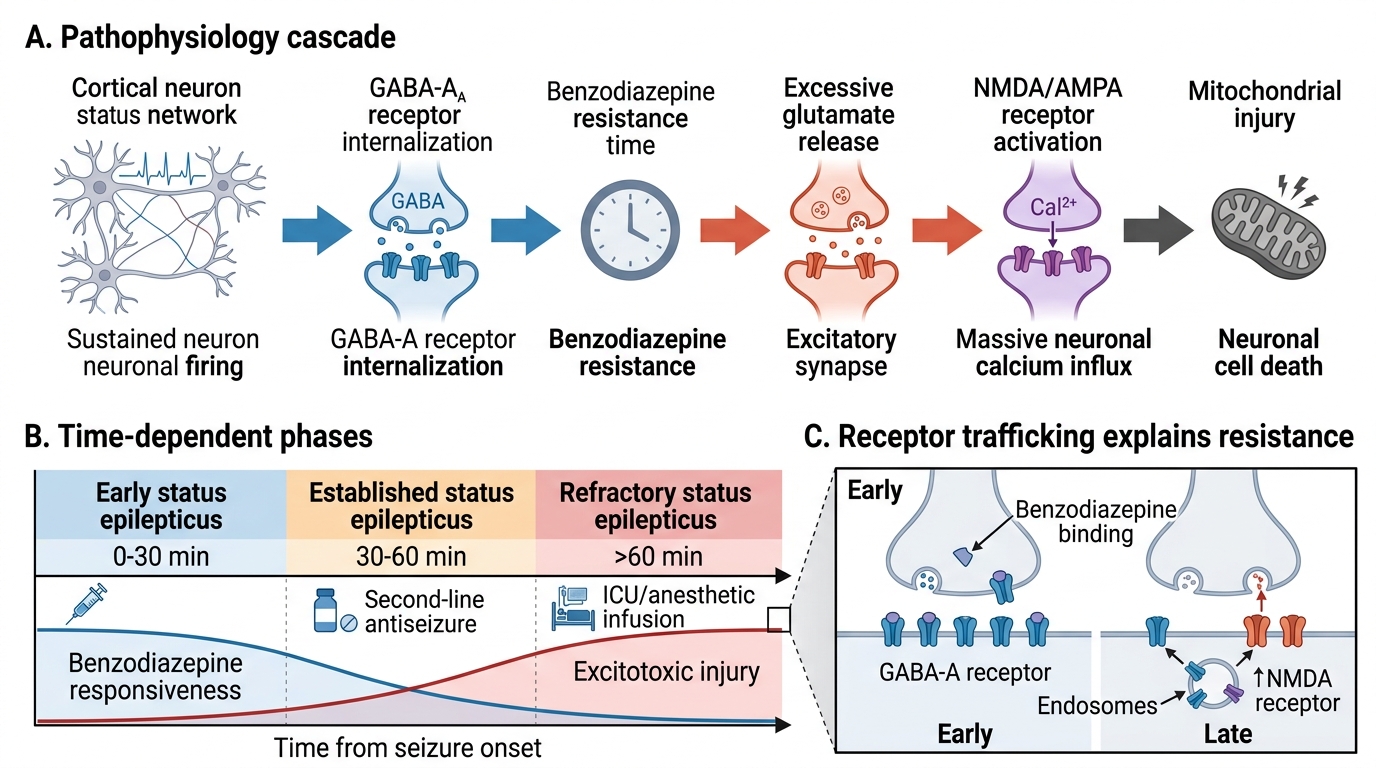
Status epilepticus (SE) is defined by the International League Against Epilepsy (ILAE, 2015) along two time dimensions: t1 (the point at which a seizure should be treated as status epilepticus, i.e., it is abnormally prolonged and unlikely to stop spontaneously — ≥5 minutes for convulsive SE) and t2 (the point at which long-term neurological consequences are likely — >30 minutes for convulsive SE). The operational definition for emergency management is t1: if a generalised tonic-clonic seizure has been going on for ≥5 minutes, treat it as SE. The previous 30-minute definition was abandoned because seizures lasting >5 minutes almost never stop spontaneously and benzodiazepines become progressively less effective with each passing minute due to receptor internalisation.
SE may be convulsive (CSE) or non-convulsive (NCSE):
• Convulsive SE: the classic presentation — continuous rhythmic jerking movements (tonic-clonic), tonic posturing, or clonic activity, with altered or absent consciousness. The ictal activity is visible clinically.
• Non-convulsive SE (NCSE): the child appears altered, confused, staring, with automatisms (lip-smacking, repetitive hand movements) but minimal or no obvious motor activity. NCSE is underdiagnosed because it looks like encephalopathy. EEG is required to confirm. NCSE must be suspected in any child who remains persistently altered after a convulsive seizure has been treated — the motor component may have stopped while electrographic seizure activity continues.
• Subtle SE (common in neonates and post-ictal states): minimal motor manifestations — nystagmus, lip smacking, pedalling, focal clonic activity — with EEG seizure activity. Particularly underrecognised in neonates.
Aetiological classification determines subsequent workup and long-term management, though it does not change the immediate treatment algorithm:
• Febrile SE (febrile status epilepticus): the most common cause in children 1–5 years (30–40% of SE in children); triggered by fever, usually without an underlying structural lesion. A prolonged febrile seizure (>15 min = complex febrile seizure) is SE by definition.
• Acute symptomatic: identifiable acute brain insult — CNS infection (meningitis, encephalitis — must be actively excluded; lumbar puncture after CT), hypo/hyperglycaemia, hyponatraemia, hypocalcaemia, hypomagnesaemia, head trauma, stroke, hypoxia-ischaemia.
• Remote symptomatic (structural): SE in a child with a pre-existing brain lesion (cortical dysplasia, prior stroke, perinatal injury).
• Idiopathic/cryptogenic: SE in a child with known idiopathic epilepsy.
• Progressive encephalopathy: Rasmussen's encephalitis, metabolic diseases (mitochondrial, lysosomal storage).
• Toxin/drug-induced: isoniazid (depletes pyridoxine), organophosphates, theophylline, antiepileptic drug withdrawal.
FIRES (febrile infection-related epilepsy syndrome): a rare but devastating form — refractory SE following a febrile illness in a previously neurologically normal child; requires specialist management.
Pathophysiology and Timeline of Status Epilepticus
Pathophysiology and Aetiology of Status Epilepticus
The pathophysiology of status epilepticus involves a failure of the normal mechanisms that terminate seizures. Under normal conditions, a seizure begins when a local excitatory imbalance (usually triggered by a brief increase in glutamate activity or sudden GABA failure) propagates, but is self-limited by GABA-mediated inhibitory surround, depletion of excitatory neurotransmitter, after-hyperpolarisation, and adenosine release. In SE, these termination mechanisms fail — either because the initiating insult is too severe, the inhibitory reserve is depleted, or both.
The critical molecular event that underlies benzodiazepine resistance in prolonged SE is GABA-A receptor internalisation: as SE persists beyond 5–10 minutes, synaptic GABA-A receptors are internalised (endocytosed) into the cell membrane, removing them from the synapse. Since benzodiazepines act by binding to the GABA-A receptor-chloride ionophore complex and enhancing chloride conductance, their efficacy diminishes progressively as receptors disappear from the synapse. This is the molecular basis of the time-sensitivity of SE treatment — benzodiazepines given at 5 minutes are far more effective than the same dose given at 30 minutes.
Concurrently, NMDA-receptor-mediated glutamate excitotoxicity increases: sustained depolarisation leads to excessive calcium entry through NMDA channels → mitochondrial dysfunction → oxygen free-radical production → neuronal apoptosis. The brain regions most vulnerable are the hippocampus (CA1 and CA3 zones), cerebral cortex, thalamus, and Purkinje cells of the cerebellum — explaining why prolonged SE produces cognitive impairment, memory deficits, and motor sequelae.
Systemic consequences of SE compound the neuronal injury:
• Phase 1 (0–30 min, compensated): tachycardia, hypertension, hyperglycaemia (catecholamine surge), hypoxia (apnoea during ictus), lactic acidosis — but cerebral autoregulation is intact and cerebral blood flow is increased to match the metabolic demand of seizing neurons
• Phase 2 (>30 min, decompensated): cerebral autoregulation fails; hypoxia, hypoglycaemia, hyperthermia, and lactic acidosis worsen; cerebral perfusion pressure drops despite increased intracranial pressure; the brain can no longer compensate for the metabolic demands of continuous seizure activity
• Hyperthermia (core temperature >40°C) occurs in 30–50% of SE cases due to excessive muscle activity and sympathetic activation; it independently causes neuronal injury and must be treated with antipyretics and physical cooling
The aetiology determines urgency of specific interventions beyond antiepileptic drugs: CNS infection (dexamethasone before/with first antibiotic dose in bacterial meningitis to reduce hearing loss and neurological sequelae; empirical antibiotics + aciclovir for HSV encephalitis); hyponatraemia (hypertonic saline 2–4 mL/kg of 3% NaCl for symptomatic hyponatraemia-related SE); isoniazid toxicity (pyridoxine 70 mg/kg IV).
Diagnosis and Initial Investigation
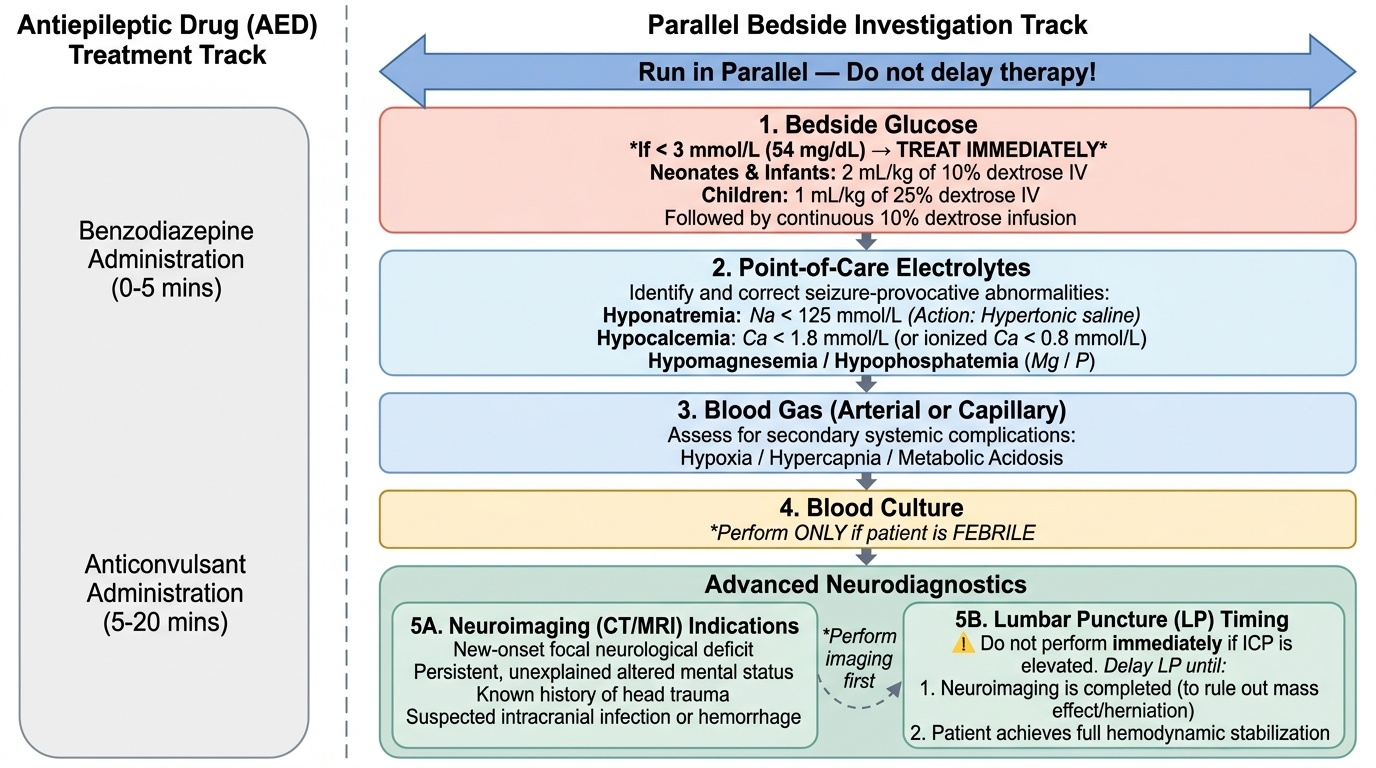
Investigation of status epilepticus occurs in parallel with treatment — antiepileptic drugs are never withheld while investigations are obtained. The investigation strategy is guided by the clinical context, prioritising the bedside tests that yield immediately actionable results (specifically, identifying treatable metabolic causes that must be corrected simultaneously with antiepileptic therapy). A prolonged seizure caused by hypoglycaemia will not respond adequately to benzodiazepines without concurrent glucose correction; a seizure from hyponatraemia requires hypertonic saline in addition to anticonvulsants. The correct mental model is to run the investigation protocol as a parallel track alongside, not after, the treatment algorithm — the team member not administering the benzodiazepine should be drawing blood for glucose and electrolytes, placing the pulse oximeter, and obtaining ECG monitoring while the prescriber calculates and administers the drug. This parallel-processing habit is what separates effective emergency team management from a sequential approach that wastes critical minutes.
Provided image
Immediate bedside investigations (obtain during Phase 0–1, before or alongside first antiepileptic dose):
• Blood glucose (first and most urgent): treat if <3 mmol/L (54 mg/dL) with IV glucose — 2 mL/kg of 10% dextrose (neonates and infants) or 1 mL/kg of 25% dextrose (children), followed by continuous 10% dextrose infusion
• Serum electrolytes (sodium, potassium, calcium, magnesium, phosphate): hyponatraemia (Na <125 mmol/L), hypocalcaemia (Ca <1.8 mmol/L total or ionised Ca <0.8 mmol/L), and hypomagnesaemia are all directly seizure-provocative and require specific correction
• Blood gas (arterial or capillary): assess hypoxia, hypercapnia, and metabolic acidosis
• Full blood count and blood culture: if fever or suspected CNS infection
• Urine toxicology screen: if no clear cause, or if toxin ingestion is possible (isoniazid, theophylline, anticonvulsant levels)
• Anticonvulsant levels: if child is on antiepileptic therapy — subtherapeutic levels are a common precipitant of SE
EEG: essential for diagnosing non-convulsive SE (NCSE) in a child who remains altered after motor seizure activity has stopped. NCSE can occur post-ictally or as the primary presentation. A portable EEG or continuous EEG monitoring is the definitive investigation. EEG shows continuous or near-continuous epileptiform discharges. Bedside EEG should be obtained urgently in any child with persistent altered mental status despite seizure control.
Neuroimaging: indications for emergency CT head in SE:
• Focal neurological deficit not explained by post-ictal paralysis (Todd's paresis)
• Failure to return to baseline within 1 hour despite clinical seizure control
• Suspicion of trauma or non-accidental injury
• First unprovoked seizure in a child >5 years with no family history
• Signs of raised intracranial pressure (fundal changes, bradycardia + hypertension + respiratory irregularity — Cushing's triad)
MRI brain (diffusion-weighted imaging) is the preferred modality when time permits — it detects hippocampal changes of SE-related injury and identifies structural causes not visible on CT.
Lumbar puncture for CSF analysis: performed after neuroimaging (to exclude mass lesion/raised ICP) and after haemodynamic stabilisation. Indicated in all febrile SE cases where CNS infection cannot be excluded. Do NOT delay antibiotics and aciclovir for LP results if meningitis/encephalitis is clinically suspected.
SELF-CHECK
A 3-year-old child (weight 14 kg) has been having a generalised tonic-clonic seizure for 7 minutes. There is no IV access. The nurse has a buccal midazolam preparation available. What is the correct dose and route?
A. 0.1 mg/kg IV (not possible without IV access)
B. 0.2 mg/kg buccally — 2.8 mg squirted into the buccal mucosa
C. 0.5 mg/kg rectally — 7 mg rectal diazepam
D. 18 mg/kg IV phenytoin — second-line drug
Reveal Answer
Answer: B. 0.2 mg/kg buccally — 2.8 mg squirted into the buccal mucosa
When IV access is not available, midazolam buccal (or IM) at 0.2–0.3 mg/kg is the first-line treatment of choice. For a 14 kg child: 0.2 mg/kg × 14 = 2.8 mg buccally (the standard commercially available midazolam buccal formulation is 5 mg/mL; give ~0.6 mL into the buccal mucosa). Lorazepam 0.1 mg/kg would require IV access. Rectal diazepam 0.5 mg/kg (= 7 mg) is an alternative when neither IV access nor midazolam buccal preparation is available. Phenytoin is second-line, not first-line, and requires IV access.