Page 10 of 34
PE26.4 | Hemolytic Anaemia — SDL Guide
Learning Objectives
- Describe the cardinal clinical triad of haemolytic anaemia and distinguish acute from chronic haemolysis
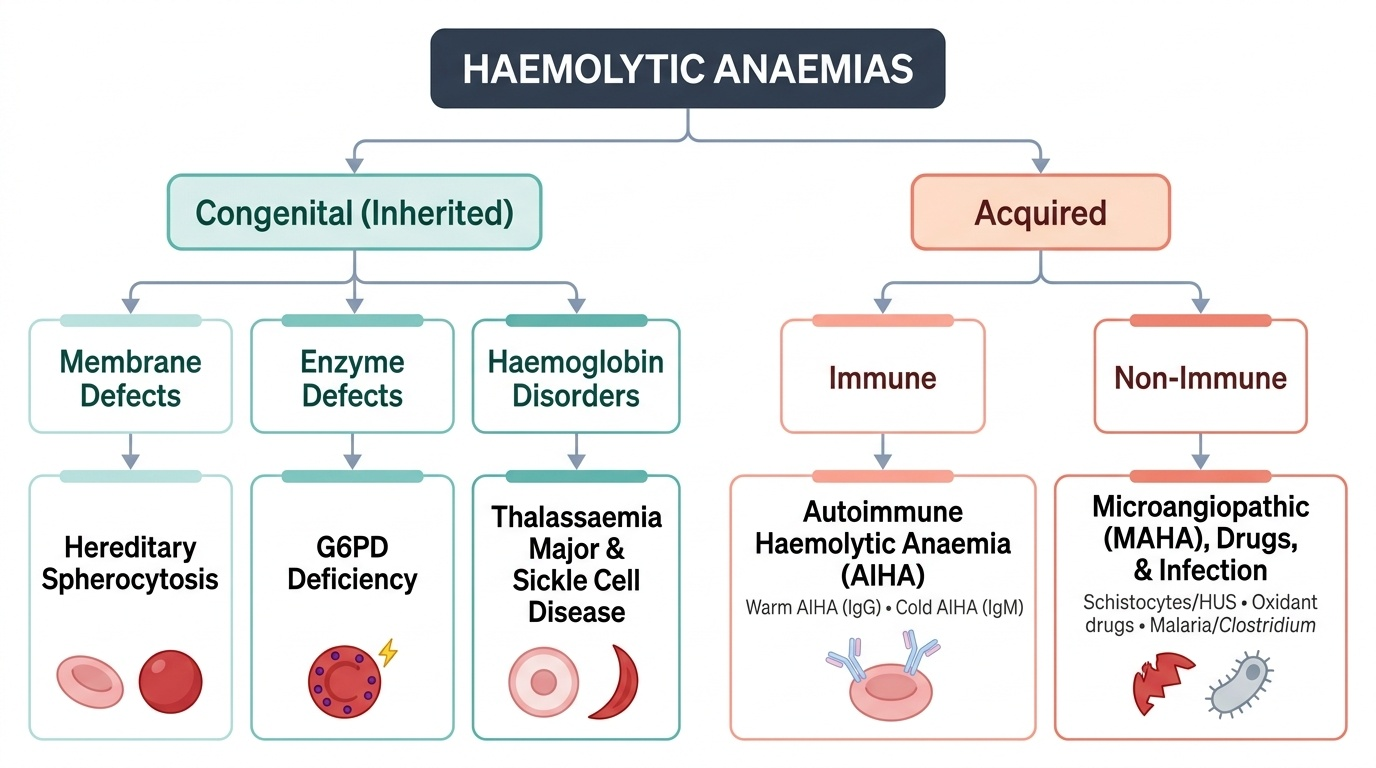
- Classify haemolytic anaemias into congenital (membrane, enzyme, haemoglobin disorders) and acquired (immune, non-immune) categories
- Explain the pathophysiology and identify the characteristic clinical and smear findings for G6PD deficiency, thalassaemia major, sickle cell disease, hereditary spherocytosis, and AIHA
- Interpret laboratory investigations — including reticulocyte count, peripheral smear, LDH, haptoglobin, Coombs test, and condition-specific tests — to confirm haemolysis and identify the cause
- Outline condition-specific management including transfusion and chelation for thalassaemia major, trigger avoidance for G6PD, splenectomy timing for HS, hydroxyurea and prophylaxis for SCD, and corticosteroids for AIHA
INSTRUCTIONS
Haemolytic anaemias represent a diverse and clinically important group of disorders in which the lifespan of red blood cells is shortened. In India, genetic haemolytic conditions — thalassaemia major, sickle cell disease, and G6PD deficiency — are among the most prevalent inherited disorders and are the subject of national carrier-screening and disease-management programmes. Each condition has a distinct mechanism, a characteristic peripheral smear, and a condition-specific management protocol that you must know precisely. This module builds a systematic approach to the haemolytic anaemias by working from the shared clinical and laboratory features of haemolysis down to the distinguishing features of each major condition.
References
- Ghai Essential Pediatrics, 9th ed, Ch 17 (textbook)
- Nelson Textbook of Pediatrics, 21st ed, Ch 489–495 (textbook)
- Thalassaemia International Federation: Guidelines for the Management of Transfusion-Dependent Thalassaemia, 4th ed (guideline)
- IAP Guidelines on Sickle Cell Disease Management (guideline)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
Two children present to the same paediatric OPD on the same afternoon. Child 1 is a 4-year-old boy from a family in Odisha who has had yellow eyes and swollen abdomen for 2 years; his younger sister has similar features; CBC shows Hb 6.0 g/dL, MCV 68 fL, reticulocytes 8%. Child 2 is a 6-year-old girl who develops sudden-onset pallor, dark urine, and jaundice 3 days after eating fava beans at a relative's house; CBC shows Hb 7.0 g/dL, MCV 84 fL, reticulocytes 12%. Both have normocytic-to-microcytic anaemia with elevated reticulocytes and jaundice — the hallmark of haemolysis. But the mechanisms are entirely different, the smear findings distinct, and the management strategies nothing alike. How do you use the clinical history, ethnicity, smear, and targeted tests to separate them — and then manage each correctly?
WHY THIS MATTERS
Haemolytic anaemias are among the most clinically significant and socially impactful paediatric blood disorders in India. Beta-thalassaemia major affects over 10,000 newborns each year in India, requiring lifelong monthly transfusions and iron chelation therapy at enormous family and health-system cost. Sickle cell disease is prevalent in tribal belt states (Odisha, Chhattisgarh, Madhya Pradesh, Maharashtra) and is now covered under the National Sickle Cell Anaemia Elimination Mission 2047. G6PD deficiency affects approximately 10–15% of some Indian population groups and is the most common enzyme deficiency causing acute haemolytic crises in children — triggered by common medications (antimalarials, sulfa drugs), infections, and dietary factors (fava beans). Hereditary spherocytosis is the most common hereditary haemolytic anaemia in Northern European populations but is seen across India. Understanding these conditions equips you for the realities of paediatric inpatient wards, blood transfusion centres, and tribal-area health camps.
RECALL
Review these prerequisites:
• Haemolytic pattern on CBC: normocytic anaemia (unless concurrent deficiency) + elevated reticulocyte count (>2%) + elevated unconjugated (indirect) bilirubin + reduced haptoglobin + elevated LDH — this constellation was introduced in the Approach to Anaemia SDL.
• Haemoglobin structure (Biochemistry): HbA (normal adult) = α₂β₂; HbF (fetal) = α₂γ₂; HbS = β-chain with valine substituted for glutamic acid at position 6 (glutamic acid is negatively charged, valine is non-polar — this small substitution causes polymerisation under deoxygenation). HbF is protective in both sickle cell disease and beta-thalassaemia because it has no beta chains.
• Splenomegaly: the spleen is the primary organ of extravascular haemolysis — abnormal or antibody-coated RBCs are trapped and destroyed by splenic macrophages; chronic haemolysis leads to progressive splenomegaly and, in thalassaemia and sickle cell disease, eventual hypersplenism.
• Direct Coombs test (DAT): detects IgG or complement bound to the RBC surface; positive in autoimmune haemolytic anaemia and haemolytic disease of the newborn; negative in all the congenital haemolytic conditions (IgG is not the mechanism).
Clinical Presentation of Haemolytic Anaemia
Haemolytic anaemias share a clinical triad that reflects the consequences of shortened RBC survival: anaemia (from accelerated red cell destruction outpacing production), jaundice (from unconjugated hyperbilirubinaemia due to haem catabolism), and splenomegaly (from trapping and destroying abnormal red cells). The combination of pallor with yellow sclerae and a palpable spleen in a child should immediately raise the suspicion of haemolytic disease even before the CBC result is available. This triad distinguishes haemolytic anaemia from iron deficiency (no jaundice, no splenomegaly) and megaloblastic anaemia (mild jaundice from ineffective erythropoiesis, but minimal splenomegaly).
Provided image
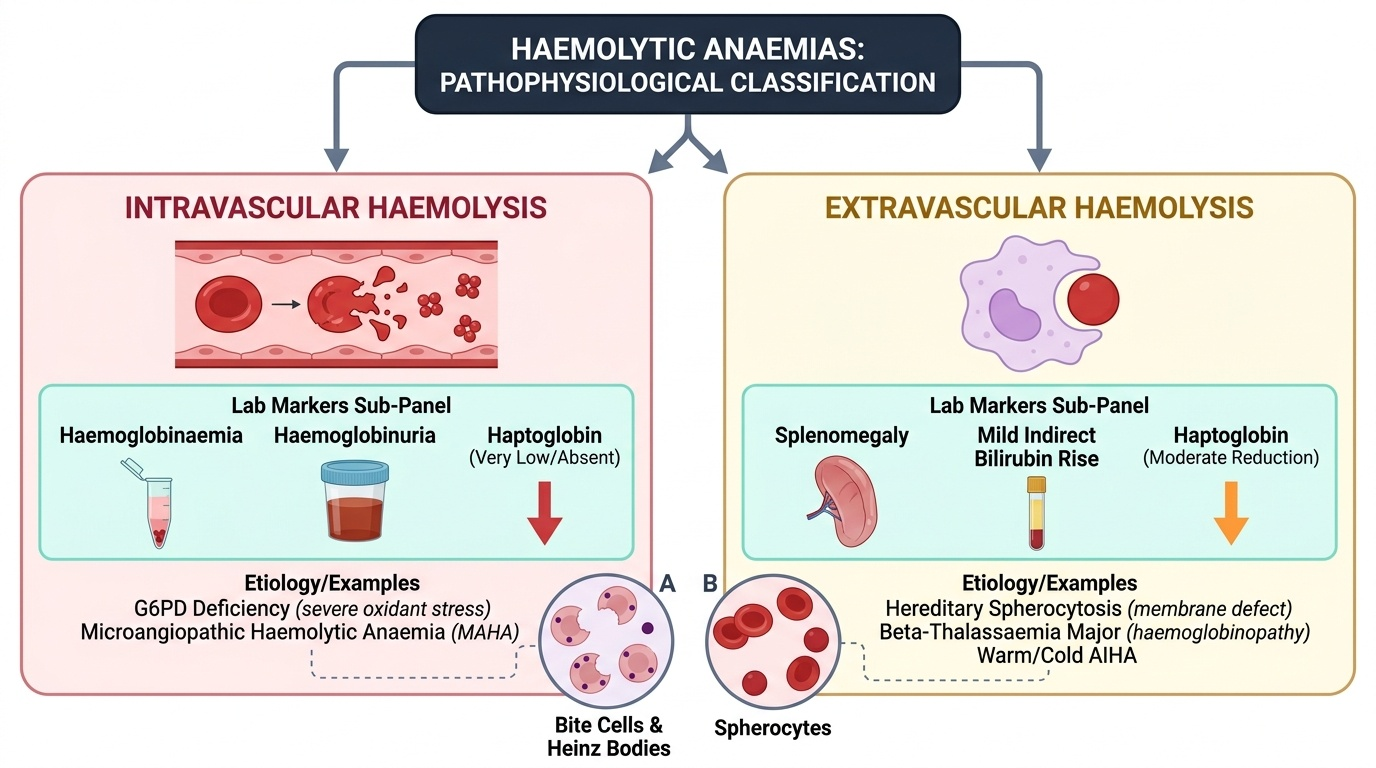
Acute haemolytic crisis: sudden-onset pallor, tachycardia, jaundice appearing within hours, dark urine (haemoglobinuria — from intravascular haemolysis), loin pain, and vomiting. In G6PD deficiency, this classic picture follows oxidant exposure (infection, drug, or fava beans) by 24–72 hours. Haemoglobinuria occurs specifically in intravascular haemolysis (complement-mediated destruction releases free haemoglobin into plasma, which is filtered into urine when haemoglobin exceeds haptoglobin binding capacity).
Chronic compensated haemolysis: in conditions with chronic haemolysis (thalassaemia major, HS, SCD between crises), the body partially compensates through erythroid hyperplasia. These children have a persistently low but stable haemoglobin, mild-moderate jaundice, and variable splenomegaly. They develop consequences of chronic haemolysis and its treatment:
• Bony changes: in thalassaemia major and untransfused/under-transfused SCD — erythroid marrow expansion into cortical bone causes facial bossing (maxillary prominence, protrusion of upper teeth — the 'thalassaemic facies'), frontal bossing, hair-on-end appearance on skull X-ray
• Gallstones (pigment): chronic bilirubin overproduction leads to calcium bilirubinate gallstones in childhood — right upper quadrant pain, elevated bilirubin, visible on USS
• Growth retardation and delayed puberty in inadequately transfused thalassaemia major
• Aplastic crisis: a temporary cessation of erythropoiesis, classically from parvovirus B19 infection (infects erythroid progenitors), causing a sudden dramatic fall in Hb with absent reticulocytes; dangerous in any chronic haemolytic condition
Pathophysiology and Classification of Haemolytic Anaemias
The pathophysiology of haemolytic anaemias divides cleanly into two major categories based on whether the defect is intrinsic to the red cell (congenital — all inherited conditions) or extrinsic to it (acquired — immune or environmental). This classification is clinically useful because it determines the diagnostic strategy and the durability of treatment. Congenital haemolytic conditions are permanent and require lifelong management strategies — trigger avoidance, chronic transfusion, or organ removal; acquired conditions may remit with treatment of the cause. Understanding why each condition produces haemolysis — the molecular mechanism — allows you to predict the smear findings, the triggering events, and the specific tests that will confirm the diagnosis, rather than memorising a disconnected list of facts. The Coombs test is negative in ALL congenital conditions, and positive only in AIHA — this single result divides the workup tree at its most important branch point.
Provided image
CONGENITAL haemolytic anaemias (intrinsic RBC defects — negative Coombs test because the mechanism is not antibody-mediated):
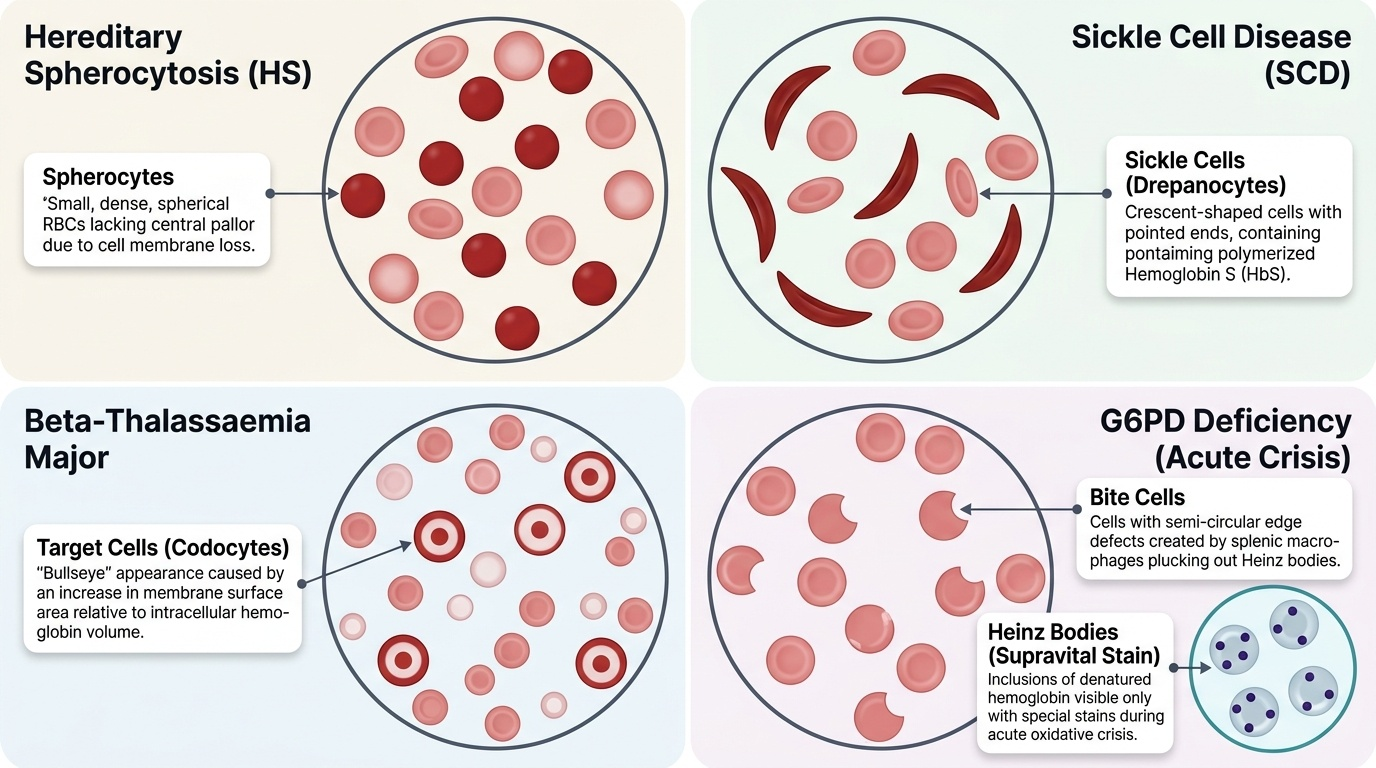
1. Membrane defects: the RBC membrane proteins (spectrin, ankyrin, band-3, protein 4.2) form a cytoskeletal scaffold that maintains the normal biconcave disc shape and deformability. In hereditary spherocytosis (HS), mutations in these proteins cause loss of membrane lipid bilayer through vesiculation — the RBC gradually shrinks and becomes a sphere (spherocyte), which is less deformable and is sequestered and destroyed by the spleen.
2. Enzyme defects: RBCs lack mitochondria and depend on glycolysis (Embden-Meyerhof pathway) for ATP and the hexose monophosphate shunt (pentose phosphate pathway) for NADPH. G6PD (glucose-6-phosphate dehydrogenase) is the first enzyme of the hexose shunt and generates NADPH, which is required to maintain glutathione in the reduced state — the cell's principal antioxidant defence. G6PD-deficient RBCs cannot neutralise oxidant stress; oxidised (denatured) haemoglobin precipitates as Heinz bodies, which attach to the inner membrane and cause the spleen to 'bite out' these inclusions, producing bite cells on smear.
3. Haemoglobin disorders (haemoglobinopathies):
- Beta-thalassaemia major: absent or markedly reduced beta-chain synthesis; alpha chains in excess precipitate within RBC precursors, causing intramedullary destruction (ineffective erythropoiesis) and haemolysis of released cells; profound anaemia develops when HbF (which does not require beta chains) declines after 6 months of age.
- Sickle cell disease (SCD): the abnormal HbS polymerises under deoxygenation, distorting the RBC into a sickle shape; sickled cells are rigid and obstruct microvascular blood flow (vasoocclusion) causing pain crises, and are haemolysed by both extravascular (spleen) and intravascular mechanisms.
ACQUIRED haemolytic anaemias (extrinsic defects — intrinsic RBC structure is normal):
1. Immune (AIHA): antibodies directed against RBC surface antigens cause complement activation or direct splenic phagocytosis; warm AIHA (IgG, most common in children) is extravascular; cold AIHA (IgM, complement-mediated) is intravascular.
2. Non-immune: microangiopathic haemolytic anaemia (MAHA) — mechanical fragmentation of RBCs in abnormal vasculature (HUS, TTP); drug-induced oxidant haemolysis; infectious haemolysis (malaria — Plasmodium directly ruptures infected RBCs; Bartonella).
Individual Conditions: G6PD, Thalassaemia, Sickle Cell, Hereditary Spherocytosis, AIHA
Each major haemolytic condition has a distinct constellation of features that allows recognition even without laboratory confirmation in many cases. Understanding these condition-specific profiles is essential for rapid triage — especially for G6PD crisis and sickle cell vasoocclusive crisis, which are paediatric emergencies.
Provided image
G6PD Deficiency:
Genetics: X-linked recessive — predominantly affects males (hemizygous); carrier females are usually unaffected but may have intermediate enzyme activity. Over 400 million individuals are affected globally; common in malaria-endemic regions including India. Trigger-dependent haemolysis is the defining feature — between episodes, haemoglobin and smear are normal; haemolysis is acute and episodic, not chronic. Common triggers: fava beans (favism — the most dramatic presentation), primaquine, dapsone, nitrofurantoin, sulfa antibiotics, infections (including COVID-19). Smear during crisis: Heinz bodies (supravital stain — not visible on Leishman stain), bite cells (RBCs with a chunk bitten out by splenic macrophages removing Heinz body inclusions), and spherocytes. G6PD enzyme assay is the diagnostic test; perform after the crisis has resolved — enzyme activity is falsely high during acute haemolysis because the G6PD-deficient older cells are destroyed and the remaining reticulocytes have higher enzyme activity.
Thalassaemia Major (Beta-Thalassaemia Major):
Genetics: autosomal recessive; both parents are thalassaemia minor (carriers); child is homozygous for beta-chain mutations. Presents after 6 months of age as HbF declines. Clinical features: severe anaemia (Hb 2–6 g/dL without transfusion), progressive massive splenomegaly and hepatomegaly, thalassaemic facies (frontal and maxillary bossing from erythroid marrow expansion), growth failure, and, without adequate treatment, death in childhood. Smear: microcytic hypochromic cells, target cells (codocytes), nucleated RBCs, marked poikilocytosis. Diagnosis: haemoglobin electrophoresis shows markedly elevated HbF (>70–90%), absent or markedly reduced HbA, elevated HbA₂. Serum ferritin is elevated (from chronic haemolysis and transfusion iron load). Pre-transfusion Hb target: ≥9–10 g/dL.
Sickle Cell Disease (SCD — HbSS):
Genetics: autosomal recessive; HbSS from inheritance of HbS from both parents. Prevalent in Odisha, Chhattisgarh, Madhya Pradesh, Maharashtra tribal populations in India. Clinical features: vasoocclusive pain crises (bone pain, acute chest syndrome, stroke), dactylitis (painful swelling of hands and feet — often the first presentation in infants, also called hand-foot syndrome), splenic sequestration crisis (sudden Hb drop + splenomegaly), aplastic crisis (parvovirus B19), and progressive functional asplenia (autosplenectomy) from repeated infarction. Smear: sickle-shaped cells (drepanocytes), target cells, Howell-Jolly bodies (nucleated RBC remnants — sign of asplenia), polychromasia. Diagnosis: haemoglobin electrophoresis (HbS predominant, absent HbA); sickling test (Na-metabisulphite screen). HbF is protective — hydroxyurea increases HbF production and reduces crisis frequency.
Hereditary Spherocytosis (HS):
Genetics: autosomal dominant in 75% of cases; de novo mutations in 25%. Clinical features: compensated haemolytic anaemia (Hb 9–11 g/dL), mild jaundice, splenomegaly, and pigment gallstones in childhood. Aplastic crisis (parvovirus B19) is a major risk. Smear: spherocytes (small, round, dense, no central pallor) — the defining morphological finding. MCHC is elevated (>35 g/dL — spherocytes have reduced surface-to-volume ratio, so Hb is more concentrated). Osmotic fragility test: spherocytes lyse at higher salt concentrations than normal RBCs (reduced tolerance to osmotic stress). EMA binding test (eosin-5'-maleimide flow cytometry) is the modern preferred diagnostic test. Splenectomy (after age 5–6 to reduce post-splenectomy sepsis risk) is curative — removes the site of spherocyte destruction; requires pre-splenectomy vaccination (pneumococcal, meningococcal, Hib) and lifelong penicillin prophylaxis post-splenectomy.
Autoimmune Haemolytic Anaemia (AIHA):
Acquired; IgG-mediated (warm AIHA, most common in children) or IgM-mediated (cold AIHA). Can be primary (idiopathic) or secondary (SLE, lymphoma, infection — Mycoplasma, EBV). Smear: spherocytes, polychromasia (reticulocytes), +/− agglutination (cold type). Direct Coombs test (DAT) is positive — the diagnostic test; detects IgG or complement on the RBC surface. This is the key distinguishing feature from all congenital haemolytic conditions where DAT is negative. Treatment: warm AIHA — oral prednisolone 1–2 mg/kg/day; taper over 4–6 weeks; second-line options include rituximab, splenectomy (for refractory cases).
⚑ AI image — pending faculty review (auto-QA score 7/10; best of 3 attempts)
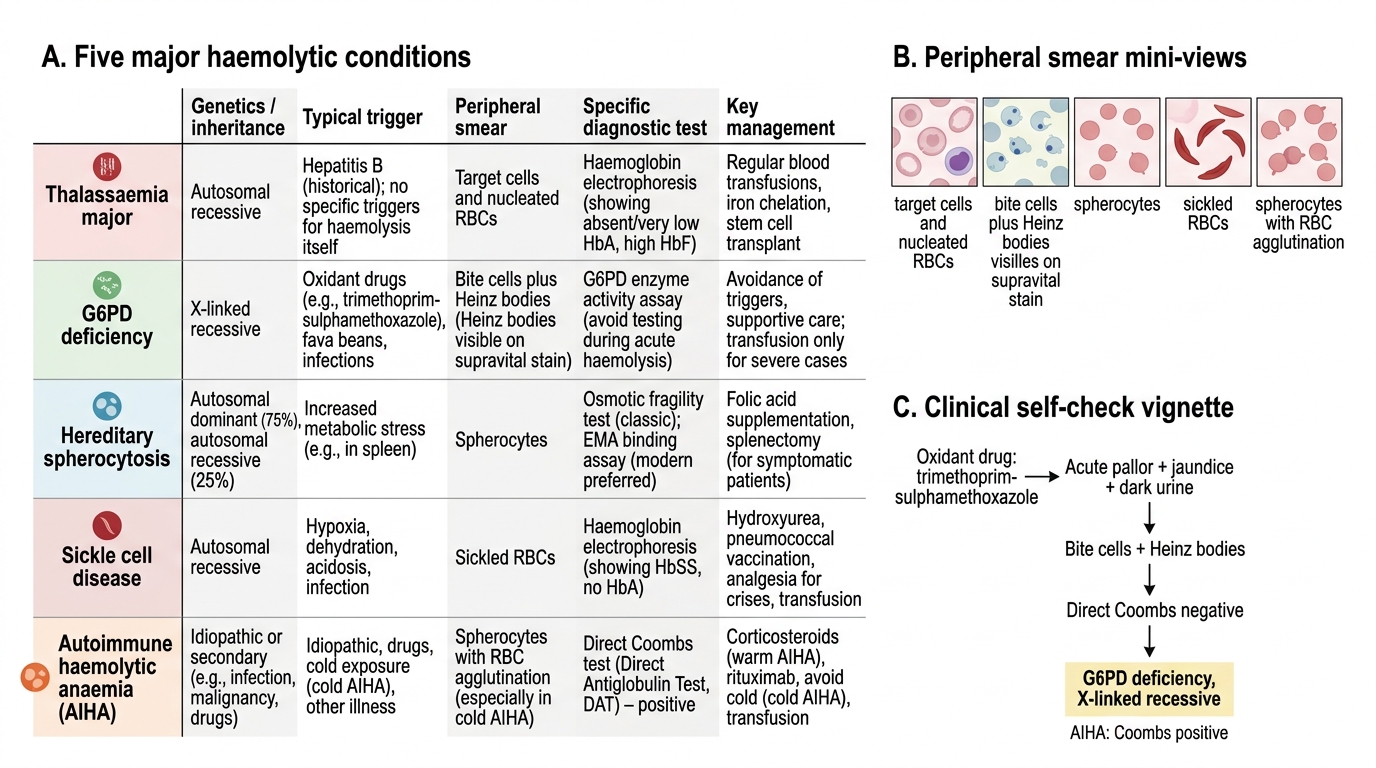
Comparison of Five Major Haemolytic Conditions
SELF-CHECK
A 5-year-old boy develops acute pallor, jaundice, and dark urine 2 days after starting trimethoprim-sulphamethoxazole for a urinary tract infection. The peripheral smear shows bite cells and Heinz bodies on supravital stain. The direct Coombs test is NEGATIVE. What is the most likely diagnosis and its inheritance pattern?
A. Autoimmune haemolytic anaemia; acquired (not inherited)
B. G6PD deficiency; X-linked recessive
C. Hereditary spherocytosis; autosomal dominant
D. Thalassaemia major; autosomal recessive
Reveal Answer
Answer: B. G6PD deficiency; X-linked recessive
Drug-triggered (sulphamethoxazole is an oxidant drug) acute haemolysis with bite cells, Heinz bodies, and a NEGATIVE Coombs test is classic G6PD deficiency. G6PD is X-linked recessive — predominantly affects males. AIHA is Coombs positive. HS shows spherocytes, not bite cells, and is not trigger-dependent. Thalassaemia major presents as chronic anaemia with transfusion dependence, not acute drug-triggered crisis, and shows target cells/nucleated RBCs.