Page 37 of 48
PE27.12 | Duchenne Muscular Dystrophy — SDL Guide
Learning Objectives
- Describe the X-linked recessive inheritance and molecular pathogenesis of DMD including the role of the dystrophin gene
- Recognise the clinical presentation: age of onset 3–5 years, proximal lower limb weakness, Gower's sign, calf pseudohypertrophy
- Explain the markedly elevated CK and its diagnostic significance; outline the role of genetic testing and muscle biopsy
- Distinguish DMD from Becker muscular dystrophy based on mutation type, dystrophin level, and clinical severity
- Outline the management approach: corticosteroids (weight-based dosing), cardiac monitoring, respiratory support, gene-targeted therapies, and family counselling
INSTRUCTIONS
Duchenne muscular dystrophy (DMD) is the most common and most severe childhood muscular dystrophy, affecting approximately 1 in 3,500 male live births. It is a relentlessly progressive disease in which boys lose ambulation in their early teens and face life-threatening cardiac and respiratory complications in young adulthood. The last decade has seen remarkable advances — corticosteroids delay disease progression, exon-skipping therapies are approved for specific mutations, and gene therapy trials are underway. Understanding DMD therefore encompasses classical genetic medicine (X-linked recessive, carrier detection, genetic counselling), clinical neuromuscular medicine (pattern of weakness, Gower's sign, pseudohypertrophy, CK interpretation, biopsy), and modern therapeutic principles (mutation-specific therapy). This SDL will equip you with the full clinical framework from diagnosis to management.
References
- Ghai Essential Pediatrics, 9th Edition, Ch. 18 (Neuromuscular Disorders) (textbook)
- Nelson Textbook of Pediatrics, 21st Edition, Ch. 627 (Muscular Dystrophies) (textbook)
- Birnkrant DJ et al. Diagnosis and management of DMD, Lancet Neurology 2018 (DMD Care Considerations) (guideline)
- IAP Guidelines on Neuromuscular Disorders (guideline)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
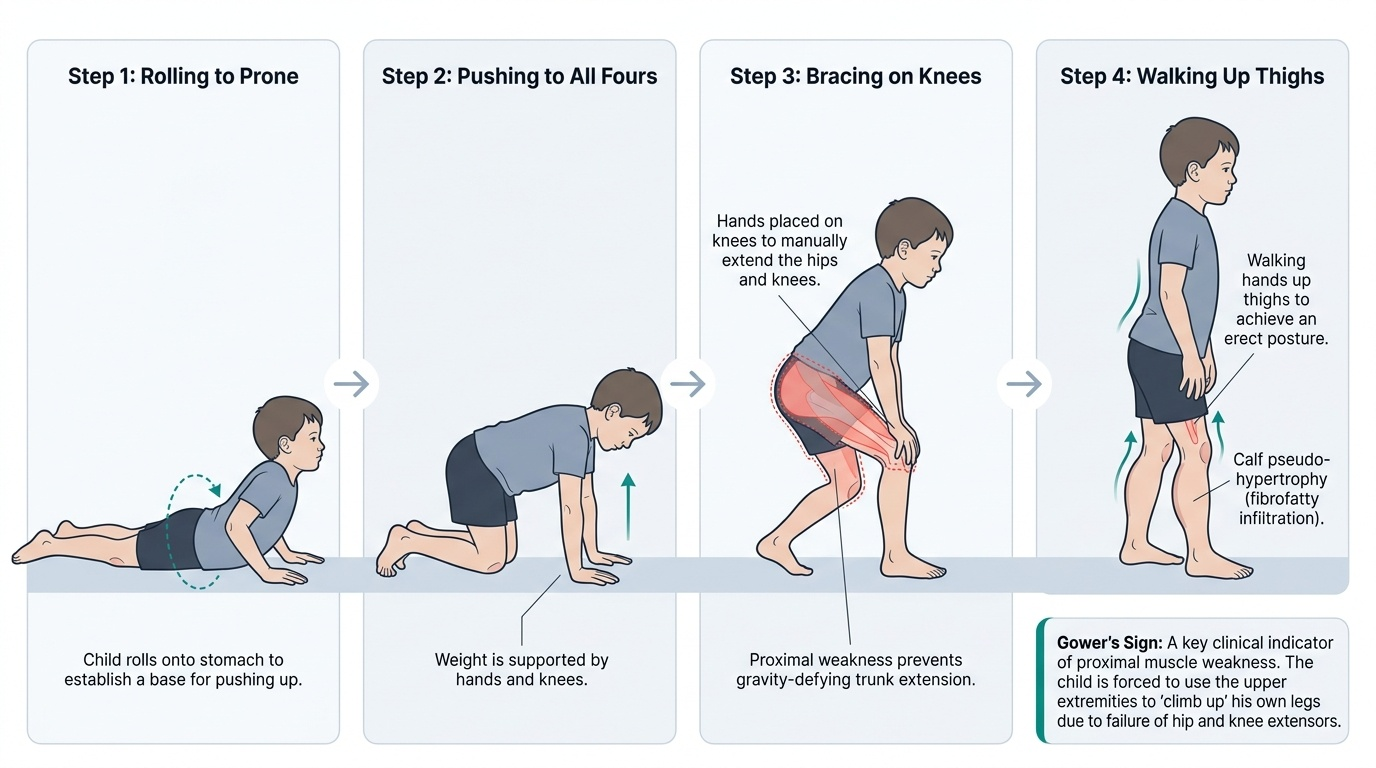
A mother brings her 5-year-old son to a paediatrics OPD with the complaint that he has been 'clumsy and slow since learning to walk.' He walks on his toes, falls frequently, and over the last six months she has noticed that he has difficulty climbing stairs and getting up from the floor — he seems to 'climb up his own legs.' On examination, he has prominent, firm calves. Neurological examination shows proximal muscle weakness (hip extensors and quadriceps grade 3/5), with normal distal strength. Deep tendon reflexes are diminished. You note the characteristic manoeuvre when you ask him to get up from the floor — he rolls onto his stomach, pushes up on all fours, then walks his hands up his thighs to achieve standing. Serum CK is 18,000 U/L (normal < 200 U/L). What is the most likely diagnosis, and what is the most appropriate next step?
WHY THIS MATTERS
Duchenne muscular dystrophy is the most common severe childhood neuromuscular disease, affecting approximately 1 in 3,500 boys globally. In India, the disease burden is substantial, with many cases diagnosed late because the characteristic presentation — toe-walking and clumsiness in an otherwise healthy-looking boy — is attributed to normal variation. Every general practitioner, paediatrician, and medical officer must be able to recognise the clinical hallmarks (proximal weakness, Gower's sign, calf pseudohypertrophy) and request the pivotal investigation (serum CK) that flags the diagnosis within minutes of the encounter. Early diagnosis enables genetic counselling for the family, carrier detection in female relatives, prenatal diagnosis for future pregnancies, timely corticosteroid therapy to extend ambulation, and proactive cardiac and respiratory monitoring. With emerging mutation-specific gene therapies, a diagnosis made at age 4–5 versus 8–10 has real consequences for the therapeutic window available to that child.
RECALL
Activate your foundational knowledge:
• X-linked recessive inheritance: males (XY) are affected (only one X); females are usually carriers (XX — one normal allele compensates). Carrier females are usually asymptomatic but can have mildly elevated CK and occasionally manifest weakness (manifesting carriers). Risk: 50% of sons affected, 50% of daughters carriers.
• Proximal vs distal weakness: proximal weakness (hip, shoulder girdle, thigh) presents as difficulty rising from the floor, climbing stairs, and lifting arms. Distal muscles (hands, feet) are relatively preserved initially in dystrophies, which contrasts with neuropathies (which often affect distal muscles first).
• Creatine kinase (CK/CPK): a muscle enzyme; markedly elevated when there is muscle fibre necrosis. Normal <200 U/L; in DMD, typically 10–100x upper limit of normal (ULN) — values of 10,000–50,000 U/L are typical in the early ambulatory phase.
• Muscle histology: normal muscle = uniform fibre size, peripheral nuclei. Dystrophic muscle = necrosis, regeneration (central nuclei, basophilic fibres), fibrosis, fatty infiltration — most pronounced in DMD.
• Dystrophin: a large structural protein anchoring the actin cytoskeleton to the extracellular matrix via the dystrophin-glycoprotein complex (DGC); absent in DMD, reduced/truncated in Becker MD.
Clinical Presentation: Recognising Duchenne Muscular Dystrophy
Duchenne muscular dystrophy (DMD) typically presents between the ages of 3 and 5 years, when increased motor demands begin to expose the underlying proximal muscle weakness. The child is usually described as having been 'clumsy from the time he started walking' — parents may notice frequent falls, difficulty running, toe-walking (from Achilles tendon tightening due to lower limb spasticity-mimic from muscle imbalance), and inability to keep up with peers on the playground. The onset is insidious: the early toddler period may appear relatively normal, but by school age, the pattern of weakness becomes unmistakable.
Provided image
Key clinical features at presentation:
Proximal lower limb weakness: The earliest and most prominent feature. Hip extensors and quadriceps are first affected — the child cannot rise from the floor without using his arms, cannot climb stairs without holding a rail, and cannot jump or hop effectively. The shoulder girdle (deltoids, biceps, triceps, pectoralis) is affected later, typically by 7–10 years.
Gower's sign: The pathognomonic clinical sign of proximal lower limb weakness. When asked to rise from the floor, the child rolls from supine to prone, pushes onto all fours, then progressively walks his hands up his thighs (using his arms as levers against his own legs) to achieve standing. This manoeuvre reflects inability of the hip extensors and quadriceps to extend the hip and knee against gravity without an assist. Gower's sign is not specific to DMD (any cause of severe proximal lower limb weakness can produce it) but is classically associated.
Calf pseudohypertrophy: The calves (and sometimes deltoids) appear enlarged and feel rubbery or firm. This is NOT true muscle hypertrophy — it represents replacement of normal muscle fibres with fat and connective tissue (fibrofatty infiltration), which increases bulk without functional strength. In contrast to the weak proximal muscles, the enlarged calves appear paradoxically strong on inspection but are functionally weak.
Toe-walking (equinus gait): Progressive Achilles tendon contracture due to ongoing muscle imbalance causes the child to walk on toes; this evolves as the disease progresses.
Other features: Lumbar lordosis (compensatory for hip extensor weakness), waddling gait (Trendelenburg), frequent falls, delayed motor milestones noted in retrospect, speech and learning difficulties (intellectual involvement in ~30% — mild, non-progressive cognitive component due to short dystrophin isoform expression in brain), cardiomyopathy (dilated, eventually universal).
Pathogenesis: Dystrophin Gene, Protein, and Muscle Degeneration
The molecular and cellular pathogenesis of DMD represents one of the most compelling examples in clinical medicine of how a single gene mutation translates into progressive, life-limiting organ failure through a precisely defined mechanistic chain. The dystrophin protein plays a fundamental structural role at the muscle cell membrane, and its absence sets in motion a cascade of pathological events — membrane fragility, calcium influx, enzymatic necrosis, exhaustion of regenerative capacity, and eventually fibrofatty replacement of functional muscle — that unfolds over years from an apparently normal early childhood into devastating adolescent-onset disability. Understanding this pathogenic chain is not merely academic: each step in the cascade represents a potential therapeutic target, and the gene-targeted therapies now available (exon skipping, stop-codon readthrough, gene replacement) directly interrupt the molecular defect at its source. The clinician who understands the pathogenesis can explain the rationale for specific mutation testing, interpret why some deletions cause severe DMD while others cause mild Becker MD, and counsel families about which therapies their child may be eligible for based on their specific mutation.
Provided image
The dystrophin gene:
Dystrophin (Xp21) is the largest known human gene — spanning approximately 2.4 Mb of genomic DNA with 79 exons. Its large size makes it inherently mutation-prone. Mutations causing DMD include:
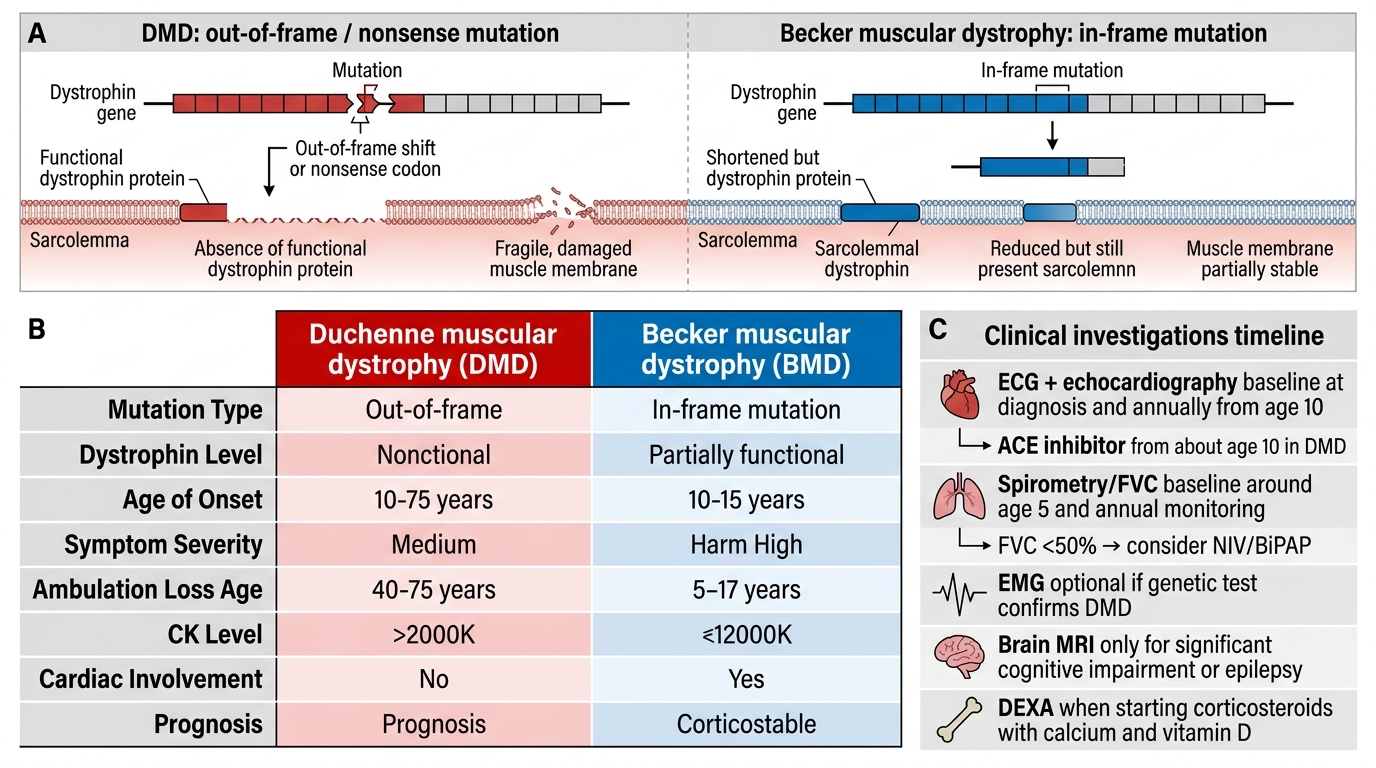
• Deletions (~65% of cases): Most commonly involving exons 45–55 or exons 2–20. Whether a deletion causes DMD or Becker MD depends on the reading frame: out-of-frame (frameshift) deletions disrupt the downstream sequence, producing a premature stop codon and absent (or markedly reduced) dystrophin → DMD. In-frame deletions allow a truncated but partially functional protein → Becker MD.
• Duplications (~10%): Also analysed by reading frame.
• Point mutations including nonsense (stop) mutations (~13%): Premature stop codon → absent protein → DMD (these are targets for ataluren/stop-codon readthrough therapy).
Dystrophin protein and the DGC:
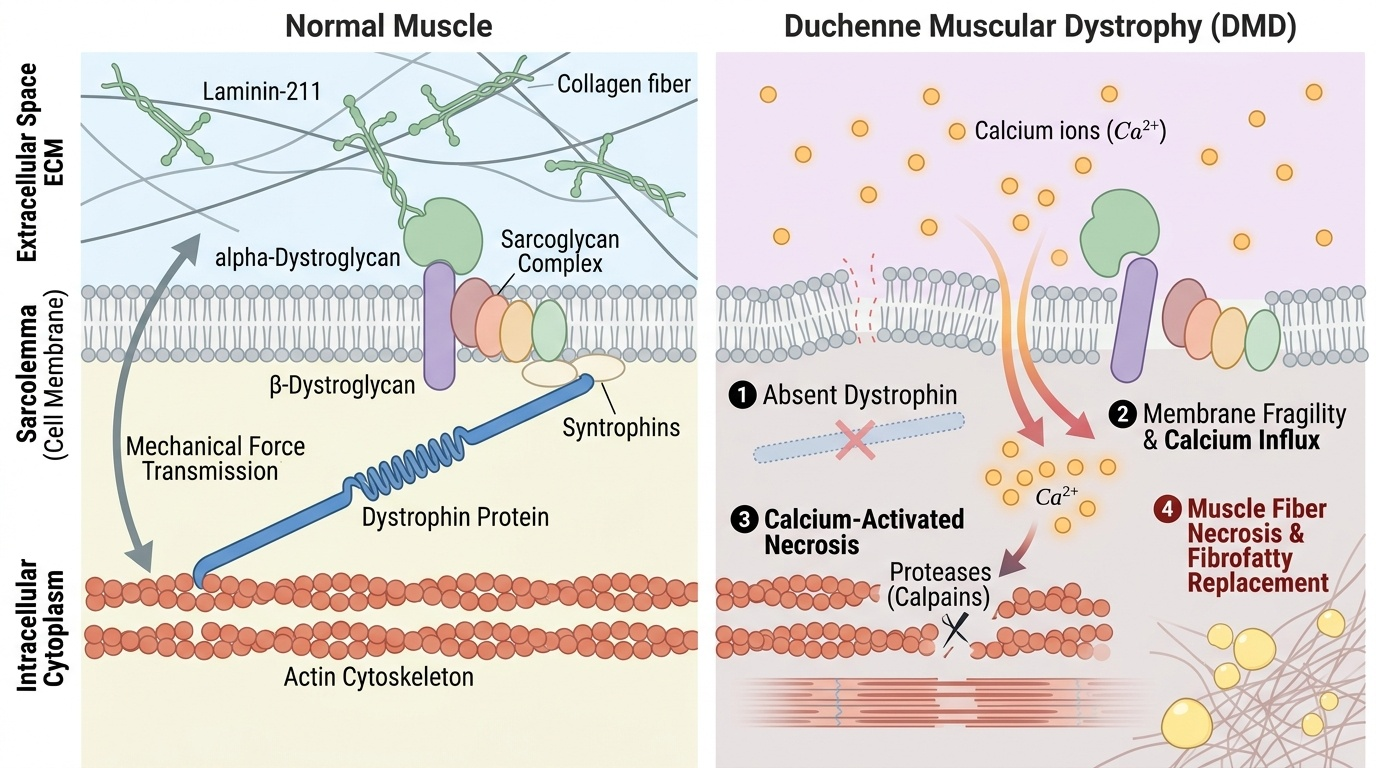
Dystrophin is a rod-shaped structural protein located at the cytoplasmic face of the sarcolemma (muscle cell membrane). It forms the core of the dystrophin-glycoprotein complex (DGC), which mechanically links the intracellular actin cytoskeleton to the extracellular matrix (laminin) through a chain of transmembrane glycoproteins. This linkage acts as a molecular shock absorber, protecting the sarcolemma from the mechanical shear forces of muscle contraction.
Absence of dystrophin → membrane fragility:
Without dystrophin, the DGC is disrupted and the sarcolemma becomes mechanically unstable. During contraction, micro-tears allow pathological influx of extracellular calcium (Ca2+) → activation of calcium-dependent proteases (calpains) → muscle fibre necrosis. The regenerative capacity of satellite cells is initially adequate (explaining the near-normal early childhood period) but becomes overwhelmed as necrosis outpaces regeneration. Progressive fibrofatty replacement of muscle fibres ensues, producing the characteristic histological findings: necrosis, regenerating fibres (central nuclei, basophilic), fibrosis, and fatty infiltration.
Cardiac involvement:
Dystrophin is also expressed in cardiac muscle. Cardiomyopathy (dilated type) is nearly universal in DMD by the late teens — it is now the leading cause of death in the post-steroid era, as respiratory management has improved survival. Cardiac monitoring and prophylactic ACE inhibitor therapy are essential from approximately 10 years of age.
SELF-CHECK
A boy with a 10-exon deletion in the dystrophin gene presents at age 3 with calf pseudohypertrophy and Gower's sign. His CK is 25,000 U/L. Genetic analysis reveals the deletion is OUT-OF-FRAME (frameshift). Which statement about his likely diagnosis and the significance of the reading frame is CORRECT?
A. He has Becker MD — out-of-frame mutations produce a truncated but functional protein
B. He has DMD — out-of-frame (frameshift) mutations disrupt downstream sequence, producing a stop codon and absent dystrophin
C. Reading frame does not matter — any deletion causes the same phenotype regardless of whether it is in-frame or out-of-frame
D. He has DMD — all deletions in exons 45–55 always cause DMD regardless of reading frame
Reveal Answer
Answer: B. He has DMD — out-of-frame (frameshift) mutations disrupt downstream sequence, producing a stop codon and absent dystrophin
The reading frame rule is fundamental to DMD vs Becker distinction: OUT-OF-FRAME (frameshift) deletions disrupt the downstream coding sequence, causing a premature stop codon and absent/non-functional dystrophin → severe DMD phenotype. IN-FRAME deletions preserve the reading frame, allowing production of an internally deleted but partially functional truncated dystrophin → milder Becker MD phenotype. This is the molecular basis of exon-skipping therapy (converting an out-of-frame to an in-frame deletion). The reading frame rule predicts phenotype in ~90% of cases.
Diagnosis and Investigations
The diagnostic workup of DMD follows a stepwise approach: an elevated CK raises the suspicion → genetic testing confirms the dystrophin mutation → muscle biopsy is reserved for cases where genetics are inconclusive. Cardiac and respiratory assessments are initiated at diagnosis as baseline evaluations for comorbidity monitoring.
Serum CK (creatinine kinase):
The single most valuable initial investigation. In DMD, CK is markedly elevated — typically 10 to 100 times the upper limit of normal (ULN) — values in the range of 5,000–50,000 U/L (ULN ~200 U/L) are characteristic in the early ambulatory phase. CK is highest when muscle mass is greatest (early ambulatory phase) and paradoxically declines in later non-ambulatory stages as muscle has been replaced by fibrous and fatty tissue. CK in Becker MD is also elevated but generally less markedly so and peaks later. A CK greater than 10 times normal in a boy with proximal weakness should prompt immediate genetic testing for DMD.
Genetic testing (FIRST-LINE confirmatory test):
• MLPA (Multiplex Ligation-dependent Probe Amplification): Detects deletions and duplications in the dystrophin gene — identifies ~75% of DMD-causing mutations. This is the first-line genetic test.
• Next-Generation Sequencing (NGS) panel / Whole-exome sequencing: For remaining point mutations (nonsense, missense, splice site) not detected by MLPA.
• Genetic confirmation establishes the specific mutation → guides mutation-specific therapy eligibility (exon skipping, stop-codon readthrough) and enables carrier testing and prenatal diagnosis.
Muscle biopsy (now second-line):
Indicated when genetic testing is non-diagnostic. Immunohistochemistry for dystrophin protein (using antibodies to N-terminal, C-terminal, and rod domain epitopes) shows absent staining in DMD and reduced/truncated staining in Becker MD. Additional stains (haematoxylin-eosin for necrosis/regeneration; Gomori trichrome for fibrosis) confirm dystrophic pattern. Dystrophin Western blot quantifies protein size and abundance.
⚑ AI image — pending faculty review (auto-QA score 4/10; best of 3 attempts)
Duchenne vs Becker Muscular Dystrophy
Additional investigations:
• ECG and Echocardiography: Baseline at diagnosis; annually from age 10; DMD-associated dilated cardiomyopathy may precede symptoms (start ACE inhibitor prophylactically from ~10 years).
• Spirometry (pulmonary function tests — FVC): Baseline when child can cooperate (~5 years); annual monitoring; FVC <50% triggers consideration of non-invasive ventilation (NIV/BiPAP).
• EMG: May show myopathic pattern but is not required when genetic test confirms DMD.
• Brain MRI: If significant cognitive impairment or epilepsy.
• Bone density (DEXA): Relevant when starting corticosteroids (osteoporosis risk); calcium and vitamin D supplementation with steroids.