Page 34 of 48
PE27.11 | Floppy Infant — SDL Guide
Learning Objectives
- Define hypotonia and describe its clinical recognition using bedside manoeuvres (traction, vertical, horizontal suspension)
- Distinguish central (brain/UMN) from peripheral (LMN/NMJ/muscle) hypotonia using clinical features
- Enumerate the important causes of floppy infant including SMA, Down syndrome, Prader-Willi, HIE, and metabolic disorders
- Outline the stepwise investigation of a floppy infant including CK, EMG/NCS, SMN1 genetic testing, MRI brain, and karyotype
- Summarise management principles including supportive care and specific therapies such as nusinersen for SMA
INSTRUCTIONS
The 'floppy infant' — a neonate or young infant with generalised hypotonia — is one of the most clinically challenging presentations in paediatrics. The differential diagnosis spans the entire neuraxis from cerebral cortex to muscle, and distinguishing a life-threatening neurological emergency from a benign self-limiting condition requires a systematic anatomical approach. The ability to perform bedside hypotonia assessment manoeuvres, interpret the neurological examination in the context of the infant's alertness and reflexes, and direct investigations efficiently is a core clinical skill for every paediatrician and neonatologist. Disease-modifying therapies such as nusinersen (for SMA) have transformed prognosis for some conditions, making early and accurate diagnosis increasingly important.
References
- Ghai Essential Pediatrics, 9th Edition, Ch. 18 (Developmental Paediatrics) (textbook)
- Nelson Textbook of Pediatrics, 21st Edition, Ch. 625 (Neuromuscular Diseases) (textbook)
- SMA Diagnosis and Care Guidelines — Cure SMA International Guidelines (2018) (guideline)
- Volpe's Neurology of the Newborn, 6th Edition (textbook)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A neonatologist is called to see a 2-day-old male infant born at term who is noted to be 'very floppy.' The baby was born to a healthy mother after an uneventful pregnancy and delivery. On examination, the infant is alert and has good eye contact; he is not in respiratory distress but has a weak cry. He lies in a frog-leg posture. On traction from supine, there is marked head lag. On vertical suspension, he slips through the examiner's hands. On ventral (horizontal) suspension, he drapes over the examiner's hand like a rag doll. Importantly, deep tendon reflexes are absent at the knees and ankles. There are no fasciculations on the tongue. Serum CK is 120 U/L (normal). The neonatologist suspects a lower motor neuron cause. What are the two most important diagnoses to consider, and what single investigation would you request first?
WHY THIS MATTERS
Hypotonia in the neonate and young infant is a common presenting problem that accounts for a significant proportion of neonatal intensive care admissions and developmental paediatric referrals. The urgency of evaluation is variable — some causes (HIE, metabolic crisis, sepsis) are life-threatening emergencies requiring immediate management, while others (SMA type 1, congenital myopathies) follow a more chronic course but require urgent specialist input for respiratory and feeding support. The clinician's ability to perform a structured neurological examination, correctly classify the hypotonia as central or peripheral using bedside manoeuvres, and direct the correct investigations is the critical first step. In the current era, disease-modifying therapies for spinal muscular atrophy (SMN1 gene therapy — nusinersen, risdiplam, onasemnogene) have made early genetic diagnosis a clinical priority with direct therapeutic implications.
RECALL
Consolidate your prior knowledge before beginning:
• Muscle tone: the resting resistance of muscle to passive stretch; regulated by the gamma-motor neuron loop, corticospinal tract, cerebellum, and basal ganglia. Hypotonia = abnormally reduced tone.
• UMN vs LMN: UMN lesions (cortex, corticospinal tract) cause spasticity later but can initially present with hypotonia in neonates (hypotonic phase). LMN lesions (anterior horn, peripheral nerve, NMJ, muscle) cause flaccid hypotonia throughout.
• Anterior horn cell: lower motor neuron cell bodies in the spinal cord ventral horn; SMA = degeneration of these cells.
• Deep tendon reflexes (DTRs): brisk/normal in UMN lesions (cortex); absent or reduced in LMN lesions (anterior horn, peripheral nerve, NMJ). DTRs are one of the key bedside distinguishing features.
• Developmental milestone: neck holding ~3 months; head lag at 4 months is abnormal.
• HIE: perinatal hypoxia-ischaemia causing global brain injury — the most important cause of CENTRAL hypotonia in term neonates in India.
• Muscle biopsy and EMG: EMG/nerve conduction studies distinguish neuropathic (SMA) from myopathic (muscle disease) patterns; muscle biopsy classifies the specific pathology.
Clinical Presentation: Recognising the Floppy Infant
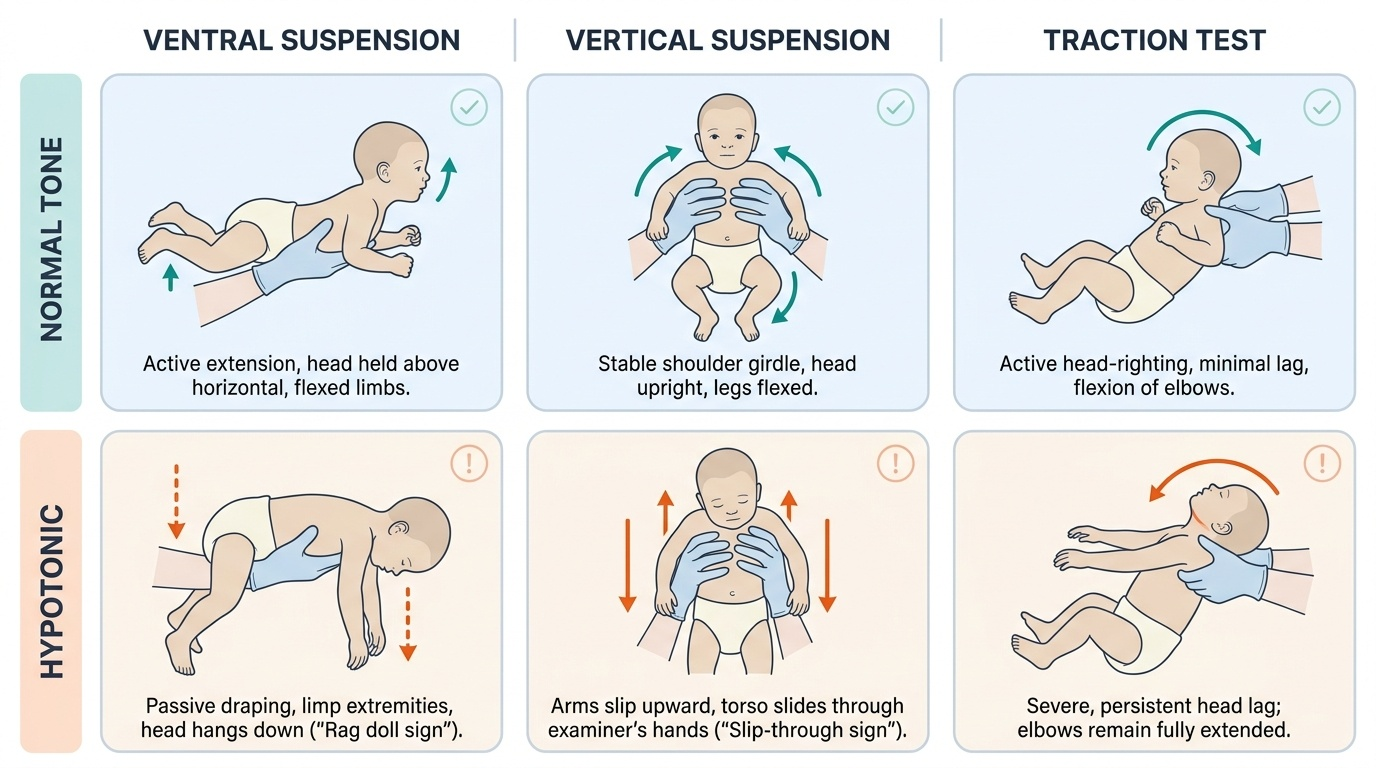
A floppy infant is one who demonstrates generalised hypotonia — abnormally reduced muscle tone — identified on examination in the neonatal or early infant period. Muscle tone is the resistance a muscle offers to passive stretch, and it is maintained by a complex reflex arc involving sensory afferents, spinal interneurons, motor efferents, and descending cortical and brainstem pathways. When any of these are disrupted, the infant fails to maintain normal anti-gravity postures and displays characteristic signs of hypotonia detectable on careful bedside examination. The diagnosis begins with four validated bedside manoeuvres that reliably quantify the degree of hypotonia and should be performed systematically on every infant with suspected motor delay or abnormal tone. These manoeuvres exploit the normal infant's ability to resist gravity with postural muscles, and compare that active resistance against the hypotonic infant's markedly diminished postural responses. Performing all four manoeuvres allows grading of hypotonia from mild to severe and provides reproducible clinical documentation for serial assessments over time.
Provided image
The four key bedside manoeuvres:
- Traction test (pull-to-sit): The examiner grasps both wrists and pulls the infant from supine to sitting. In a normal neonate, there is progressive head and trunk righting; by 4 months, no head lag. A floppy infant shows persistent, excessive head lag — the head falls completely back with no attempt to right.
- Ventral (horizontal) suspension: The infant is held prone, supported under the abdomen. A normal infant maintains a roughly horizontal position with the head raised. A hypotonic infant drapes passively over the examiner's hand, with the head, limbs, and trunk hanging limply — the 'rag doll' sign.
- Vertical suspension: The infant is held upright under the axillae. Normally, there is adequate shoulder girdle tone to resist slipping. A hypotonic infant slips through the examiner's hands ('slip-through sign') due to reduced axial and shoulder girdle tone.
- Scarf sign: The infant's arm is drawn across the chest toward the opposite shoulder; in a normal term neonate, the elbow does not reach the midline. Hypotonic infants show excessive scarf sign — the elbow crosses well past the midline, indicating reduced axial tone.
Additionally, the infant may rest in a characteristic frog-leg posture — hips abducted and externally rotated, limbs lying flat on the mattress — reflecting absent postural muscle activity.
Pathophysiology: Central vs Peripheral Hypotonia
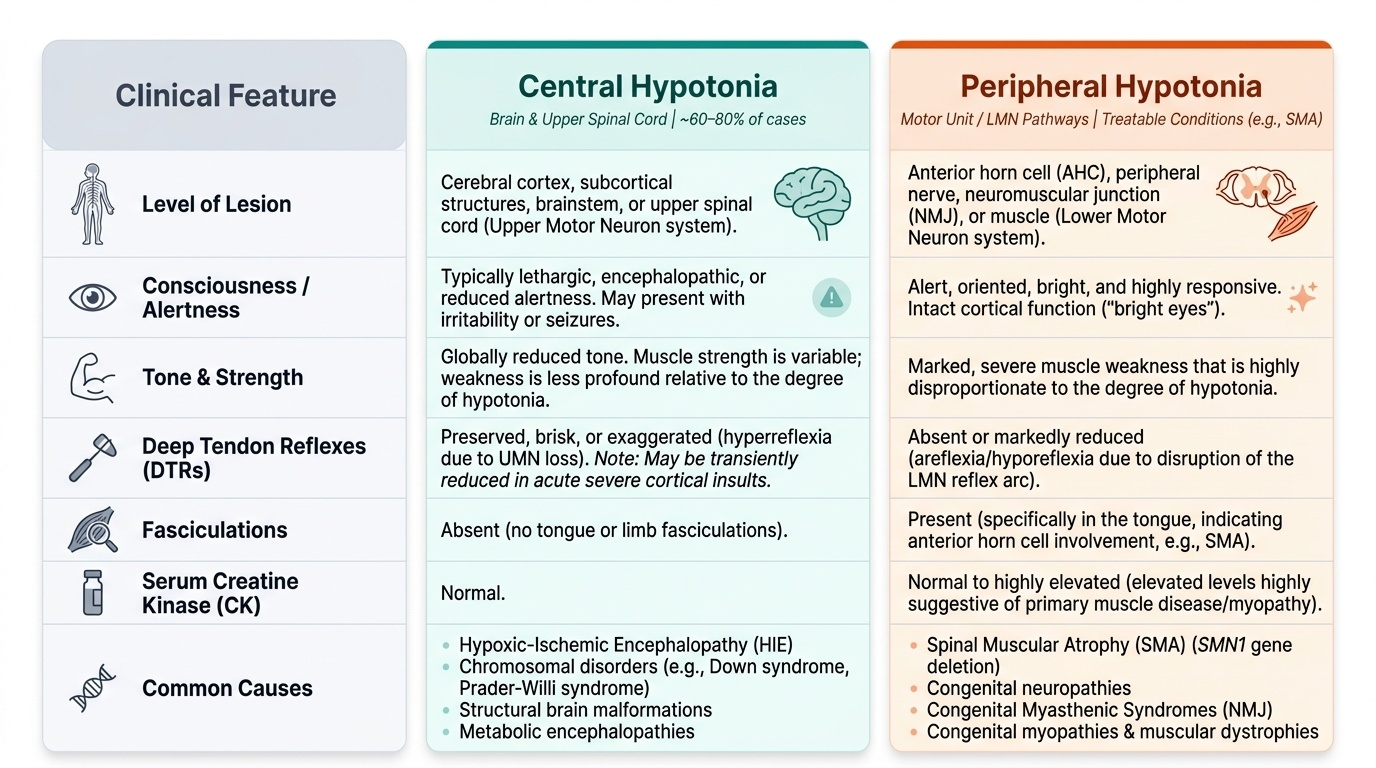
The most critical step in evaluating a floppy infant is to localise the lesion on the neuraxis. Hypotonia can arise from any level: cerebral cortex, subcortical structures, brainstem, spinal cord, anterior horn cell, peripheral nerve, neuromuscular junction, or muscle. The clinical examination differentiates these levels into two broad categories — central hypotonia (arising from the brain and upper spinal cord) and peripheral hypotonia (arising from the anterior horn cell, peripheral nerve, NMJ, or muscle). This distinction is fundamental because the causes, investigations, and management pathways diverge completely. Central hypotonia is the more common category overall (approximately 60–80% of cases), but peripheral hypotonia includes some of the most clinically important and now treatable conditions (particularly SMA).
Provided image
Central hypotonia — key features:
• The infant is typically lethargic or has reduced consciousness (encephalopathic — reduced alertness, irritability, seizures in HIE; dysmorphic or abnormal face in chromosomal/genetic causes).
• Deep tendon reflexes (DTRs) are preserved, brisk, or even exaggerated (UMN effect on spinal reflexes) — though acutely after a severe cortical insult, reflexes may be reduced initially before becoming brisk.
• Strength is variable but tone is globally reduced.
• No fasciculations in the tongue or limbs.
• Causes: HIE (commonest in India; Sarnat Grade II–III), chromosomal disorders (Down syndrome — trisomy 21; Prader-Willi syndrome — 15q11-13 paternal deletion), structural brain malformations, metabolic encephalopathies (congenital hypothyroidism, organic acidaemias), early-onset epileptic encephalopathies.
Peripheral hypotonia — key features:
• The infant is typically alert and oriented — the cortex is unaffected; bright eyes, responsive to surroundings.
• Deep tendon reflexes are absent or markedly reduced (LMN effect — loss of motor neuron or reflex arc).
• Fasciculations (rapid, involuntary rippling contractions of individual motor units, best seen in the tongue) indicate anterior horn cell involvement (SMA).
• Marked muscle weakness disproportionate to the degree of hypotonia.
• Elevated serum CK suggests muscle disease (myopathy/muscular dystrophy).
• Causes by level:
- Anterior horn cell (AHC): Spinal Muscular Atrophy (SMA) — SMN1 gene deletion, autosomal recessive; types 0–4 based on age of onset and maximum function achieved; Type 1 (Werdnig-Hoffmann) is most severe — onset before 6 months, never achieve sitting, 'frog-leg' posture, fasciculations, paradoxical breathing.
- Peripheral nerve: Congenital hypomyelinating neuropathy, Guillain-Barré syndrome (acute, post-infectious).
- Neuromuscular junction (NMJ): Neonatal myasthenia gravis (transient — passive transfer of maternal AChR antibodies, resolves in weeks), congenital myasthenic syndromes (persistent).
- Muscle: Congenital myopathies (central core, nemaline rod, myotubular), congenital muscular dystrophies, Duchenne muscular dystrophy (DMD — presentation typically later at 3–5 years but can present in infancy; see dedicated SDL PE27.12).
SMA pathogenesis: SMA results from homozygous deletion or pathogenic variants of the SMN1 (Survival of Motor Neuron 1) gene on chromosome 5q13. Loss of SMN protein leads to selective degeneration of anterior horn cells in the spinal cord and brainstem. A modifier gene, SMN2, produces a small amount of full-length SMN protein; SMN2 copy number inversely correlates with severity — more copies produce milder disease. Disease-modifying therapies (nusinersen — antisense oligonucleotide; risdiplam — SMN2 splicing modifier; onasemnogene abeparvovec — SMN1 gene replacement) work by increasing functional SMN protein production.
SELF-CHECK
A 6-week-old infant is noted to be floppy. On examination, she is alert with good eye contact, has absent knee and ankle reflexes, and visible tongue fasciculations. The mother's pregnancy and delivery were uncomplicated. Serum CK is normal. Which of the following is the most likely diagnosis?
A. Hypoxic-ischaemic encephalopathy (HIE) — central hypotonia with absent reflexes
B. Spinal Muscular Atrophy Type 1 (SMA) — anterior horn cell degeneration with absent reflexes and fasciculations
C. Congenital hypothyroidism — central hypotonia with preserved reflexes
D. Duchenne Muscular Dystrophy — muscle disease presenting with hypotonia in early infancy
Reveal Answer
Answer: B. Spinal Muscular Atrophy Type 1 (SMA) — anterior horn cell degeneration with absent reflexes and fasciculations
This infant is alert (cortex spared) with absent deep tendon reflexes and tongue fasciculations — the classic peripheral LMN pattern pointing to anterior horn cell disease. The combination of an alert infant + absent DTRs + fasciculations strongly indicates SMA Type 1 (Werdnig-Hoffmann disease). HIE would present with encephalopathy (reduced consciousness, seizures). Congenital hypothyroidism is a central cause with preserved or brisk reflexes. DMD presents at 3–5 years with proximal weakness and markedly elevated CK, not in early infancy with normal CK and fasciculations.
Differential Diagnosis and Investigation
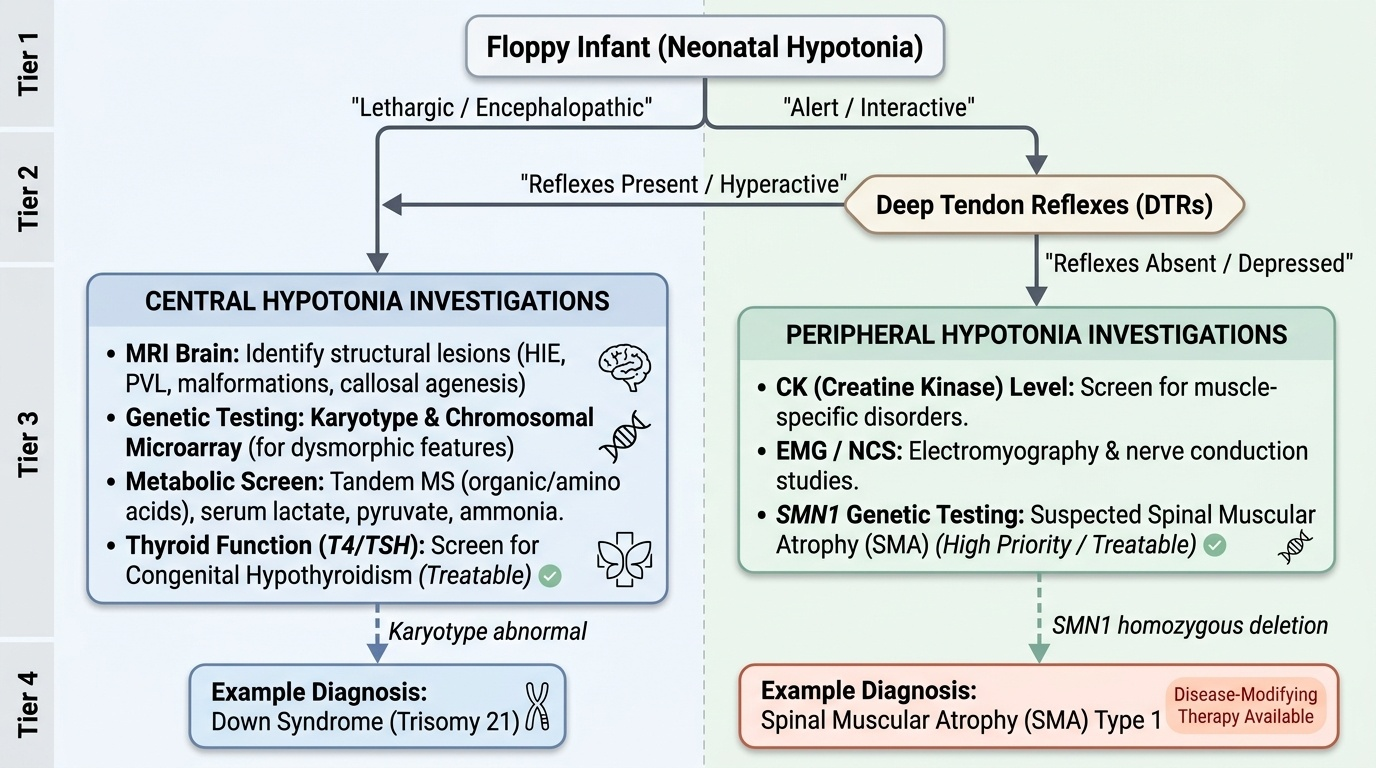
Once the clinical examination has classified hypotonia as central or peripheral using the bedside features described above, investigations are directed systematically to confirm the suspected aetiology and to exclude treatable causes that must not be missed. The approach must be guided by the clinical findings — a thorough history and meticulous examination will point to the most likely diagnosis in the majority of cases and allow investigations to be targeted rather than exhaustive. This is especially important in resource-limited settings where expensive molecular genetic testing may not be readily available. Priority is always given to conditions that are treatable — congenital hypothyroidism, neonatal myasthenia, metabolic disorders, and increasingly SMA (now that disease-modifying therapies exist) — as well as conditions with significant genetic counselling implications (autosomal recessive SMA carries a 25% sibling recurrence risk). The investigations are therefore organised by the anatomical level most likely to be responsible, not by alphabetical order or a blanket panel.
Provided image
Investigations for CENTRAL hypotonia:
• MRI brain: To identify structural lesions — HIE pattern (cortical-subcortical injury in term, PVL in preterm), cortical malformations, callosal agenesis, or metabolic white matter changes.
• Karyotype/chromosomal microarray: For dysmorphic features suggesting Down syndrome (trisomy 21), Prader-Willi (15q11 deletion/UPD — methylation analysis), or other chromosomal disorders.
• Metabolic screen: Tandem mass spectrometry (organic acids, amino acids) for metabolic encephalopathies; serum lactate/pyruvate for mitochondrial disease; ammonia (urea cycle defects).
• Thyroid function (T4/TSH): Congenital hypothyroidism — treatable; included on NBS in most Indian states.
• TORCH serology: If congenital infection suspected.
• EEG: If seizures or epileptic encephalopathy suspected.
Investigations for PERIPHERAL hypotonia:
• Serum CK: Markedly elevated (often >10x normal) in muscle diseases (DMD — typically 10–100x; other muscular dystrophies/myopathies — variable); normal or mildly elevated in SMA and neuropathies.
• Electromyography (EMG) and Nerve Conduction Studies (NCS): Distinguishes neuropathic (reduced CMAP amplitude, fibrillation potentials, positive sharp waves in SMA — denervation pattern) from myopathic (small polyphasic motor unit potentials, normal NCS) patterns.
• SMN1 genetic testing (MLPA or gene sequencing): The most important investigation when SMA is suspected — detects homozygous deletion of SMN1 exon 7 in ~95% of SMA cases. This is the first-line genetic test; muscle biopsy is NOT required if genetic testing confirms SMA.
• Muscle biopsy: For congenital myopathies (histochemistry identifies specific types — nemaline rods, central cores) and congenital muscular dystrophies (with immunohistochemistry for specific proteins — dystrophin, merosin, collagen VI).
• Anti-AChR antibodies (maternal and infant): For suspected neonatal myasthenia gravis.
Treatable causes that must not be missed:
• Congenital hypothyroidism (central hypotonia) — thyroxine replacement.
• Neonatal myasthenia gravis (peripheral, transient) — resolves with supportive care; maternal antibodies cleared by 2–8 weeks.
• SMA — nusinersen/risdiplam/onasemnogene (SMN2-targeted therapy); early treatment (pre-symptomatic or early symptomatic) dramatically improves outcomes.
• Metabolic disorders — dietary restriction (PKU, MSUD, galactosaemia).