Page 1 of 25
PA20.1 | Normal Haemostasis — SDL Guide
Learning Objectives
- Describe the vascular and platelet responses that form the primary platelet plug.
- Trace the coagulation cascade through the intrinsic, extrinsic, and common pathways, identifying which lab tests measure each arm.
- Explain thrombin's central role in haemostasis.
- Describe the cell-based model of coagulation as the physiologically accurate mental model.
- Outline the fibrinolytic system and the natural anticoagulant mechanisms that keep haemostasis in balance.
- Identify the vitamin K–dependent clotting factors and their clinical relevance.
INSTRUCTIONS
Haemostasis is the first concept that unlocks an entire cluster of bleeding and clotting disorders — platelet diseases, coagulopathies, and DIC — all taught in SDL 2–4. Understanding how the normal system works, and which lab test measures which arm of it, is the single most important foundation in haematopathology. Master this SDL before moving forward.
References
- Robbins & Cotran Pathologic Basis of Disease, 10th ed., Ch. 4 — Haemodynamic Disorders (textbook)
- Harsh Mohan Textbook of Pathology, 8th ed., Ch. 7 — Disorders of Haemostasis (textbook)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A patient with a small cut on their finger stops bleeding in 3–5 minutes. Another patient with the same cut bleeds for 45 minutes. A third patient has a normal bleeding time but develops a massive deep-tissue haematoma after a dental extraction. These three presentations point to three different arms of haemostasis — and today you will learn exactly which arm each represents, and why the lab tests for each are different.
WHY THIS MATTERS
Haemostasis is the physiological bridge between normal blood flow and pathological thrombosis or haemorrhage. Every bleeding disorder you encounter — from ITP to haemophilia A to DIC — is a failure of one component of the system you are about to study. This SDL is the foundation; SDL 2 covers platelet/vascular disorders, SDL 3 covers coagulation factor deficiencies, and SDL 4 covers DIC. Understanding the normal system now means you will not have to memorise the pathologies — you will derive them.
RECALL
Before beginning, bring to mind what you already know:
• From Year 1 Biochemistry: What does vitamin K do to clotting factors? What is the difference between a serine protease and a cofactor?
• From Year 1 Physiology: What is the difference between a vasoconstrictor response and a vasodilator response? What role do prostaglandins play in platelet behaviour?
• From Year 1 Anatomy: What is the endothelium, and why is it considered an active metabolic organ rather than just a passive lining?
Haemostasis: A Dynamic Balance

Haemostasis as a Dynamic Balance
Haemostasis is the physiological process that stops bleeding after vascular injury while keeping blood fluid within intact vessels. It is not a binary on/off switch — it is a finely regulated balance between pro-coagulant and anti-coagulant forces.
The system has three overlapping components:
1. Primary haemostasis — vascular response + platelet plug formation (seconds to ~3 minutes)
2. Secondary haemostasis — coagulation cascade → fibrin clot (minutes)
3. Fibrinolysis — dissolution of the clot once healing is complete (hours to days)
A failure at any level produces a distinct clinical and laboratory pattern. This is why understanding the anatomy of the system matters — each component has its own tests, its own diseases, and its own therapies.
Primary Haemostasis: Vascular Response
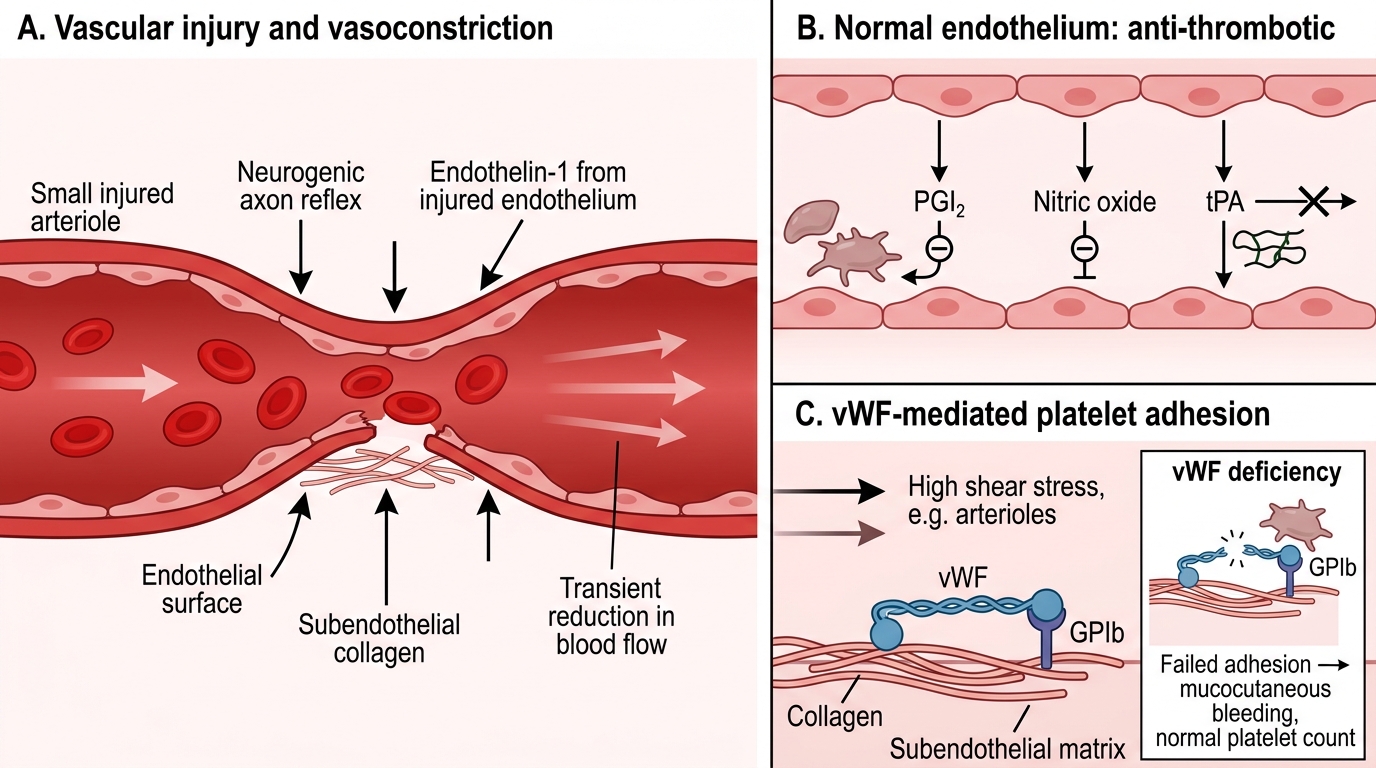
Vascular Response in Primary Haemostasis
The first response to vascular injury is vasoconstriction — neurogenic (axon reflex) and humoral (endothelin-1 released by injured endothelium). This transiently reduces blood flow, buying time for the platelet plug to form.
The normal endothelium is actively anti-thrombotic: it produces prostacyclin (PGI₂), nitric oxide, and tissue plasminogen activator (tPA), all of which inhibit platelets and promote fibrinolysis. Injury disrupts this, exposing the pro-thrombotic subendothelial matrix — particularly collagen and von Willebrand factor (vWF).
vWF is synthesised by endothelial cells and stored in Weibel-Palade bodies. At high shear stress (e.g., arterioles), vWF unfolds and bridges exposed collagen to platelet receptor GPIb — this is the initial adhesion step.
IMPORTANT: Understand that without vWF (as in von Willebrand disease), platelet adhesion fails under high shear — producing a mucocutaneous bleeding pattern identical to thrombocytopenia, but with a normal platelet count.
Primary Haemostasis: Platelet Activation and Aggregation

Primary Haemostasis: Platelet Activation and Aggregation
Once a platelet adheres via GPIb–vWF, it undergoes activation: shape change (disc → spiky sphere), degranulation, and membrane phospholipid flip.
Degranulation releases:
• α-granules: fibrinogen, vWF, P-selectin, factor V
• Dense granules (δ-granules): ADP, ATP, serotonin, calcium
Released ADP and thromboxane A₂ (TxA₂) (generated via arachidonic acid → cyclooxygenase → TxA₂) act as positive-feedback amplifiers, recruiting more platelets from the circulation.
Aggregation is the binding of recruited platelets to each other via GPIIb/IIIa receptors cross-linked by fibrinogen bridges. GPIIb/IIIa is activated by the inside-out signalling triggered by ADP/TxA₂ — this conformational change is the molecular target of clopidogrel (ADP receptor blocker) and abciximab (GPIIb/IIIa blocker).
The result is the platelet plug (primary haemostatic plug) — loose, platelet-only, adequate for minor injuries but fragile without fibrin reinforcement.

Primary Hemostasis: Platelet Adhesion, Activation, and Aggregation
Aspirin irreversibly inhibits cyclooxygenase, blocking TxA₂ synthesis. Because platelets have no nucleus (and cannot re-synthesise COX), a single aspirin dose blocks platelet function for the entire platelet lifespan (8–10 days) — clinically important for pre-operative assessment.
SELF-CHECK
A patient is found to have a normal platelet count, a prolonged bleeding time, and reduced ristocetin-induced platelet aggregation. The most likely defect is in:
A. GPIIb/IIIa receptor function
B. von Willebrand factor
C. Fibrinogen synthesis
D. Dense granule content
Reveal Answer
Answer: B. von Willebrand factor
Ristocetin induces vWF-mediated platelet agglutination — a reduced response specifically points to vWF deficiency or dysfunction (von Willebrand disease). GPIIb/IIIa deficiency (Glanzmann thrombasthenia) gives absent aggregation with ADP/collagen but normal ristocetin response. The normal platelet count rules out thrombocytopenia.