Page 3 of 21
PA16.1 | Haemolytic Anaemia — Definition, Classification & Lab Markers — SDL Guide (Part 3)
The Haemolysis Laboratory Marker Map — An Integrated View

The Hemolysis Laboratory Marker Map — Integrated Diagnostic Approach
Integrating all markers into a decision framework helps you distinguish haemolysis from other causes of anaemia and characterise its type:
Step 1 — Is there haemolysis?
Look for the haemolysis triad: ↑reticulocytes + ↑unconjugated bilirubin + ↑LDH. If all three are present, haemolysis is likely regardless of the specific cause.
Step 2 — What type?
• ↓haptoglobin + haemoglobinuria + urinary haemosiderin → intravascular haemolysis → think PNH, MAHA, G6PD crisis, ABO mismatch.
• Normal/mildly ↓haptoglobin + splenomegaly + no haemoglobinuria → extravascular haemolysis → think hereditary spherocytosis, warm AIHA, thalassaemia.
Step 3 — What is the red cell morphology telling you?
Spheres → hereditary spherocytosis or AIHA. Schistocytes → MAHA. Sickles → sickle-cell disease. Bite cells → G6PD. This step often gives the diagnosis directly.
Step 4 — Hereditary or acquired?
• Family history + lifelong symptoms + intrinsic red cell morphology → hereditary.
• Recent onset in previously well adult + autoimmune markers or microangiopathic picture → acquired.
IMPORTANT: A normal haptoglobin in a jaundiced patient with reticulocytosis should not reassure you — haptoglobin is an acute-phase reactant that can be falsely elevated during infection, masking haemolysis. Always interpret it in context.

Hemolytic Anemia: Laboratory Markers and Clinical States
The Compensated Haemolytic State in Detail
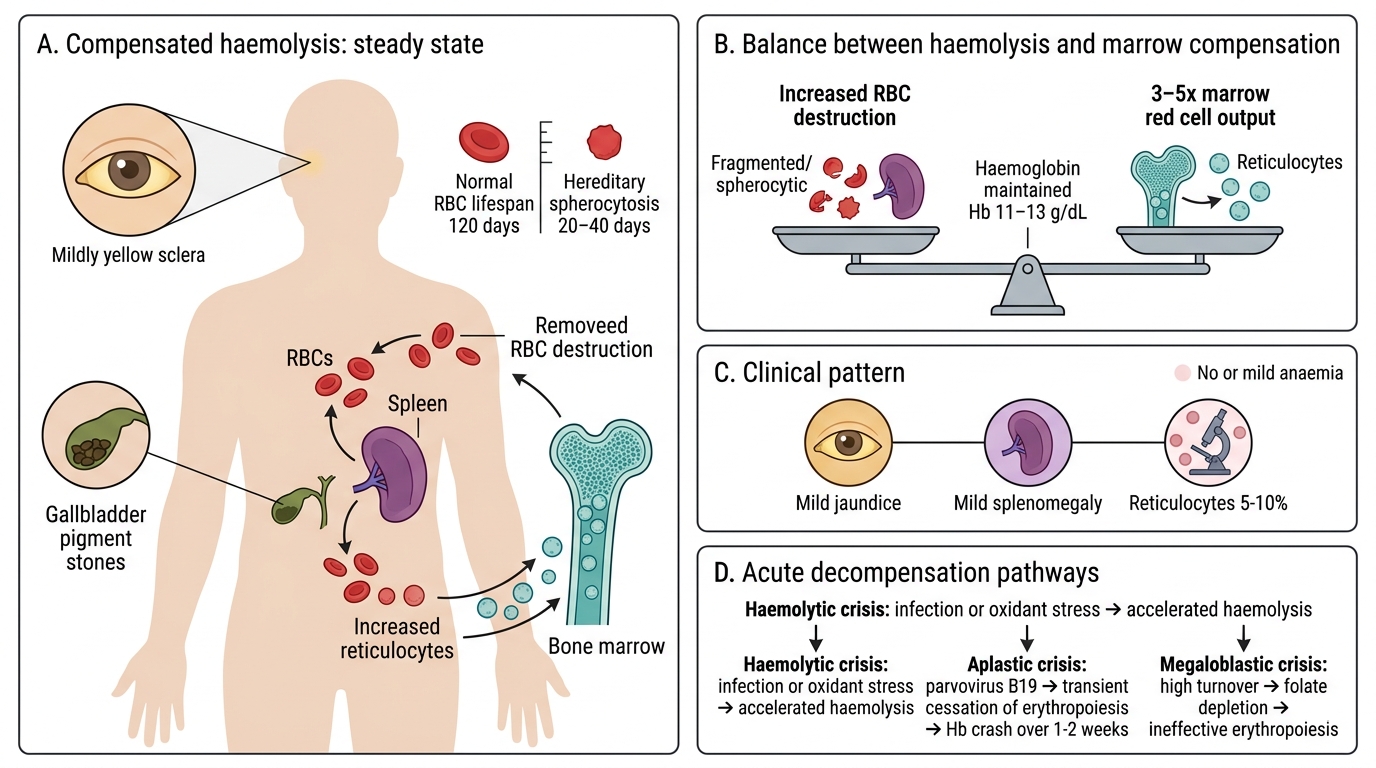
Compensated Haemolytic State
The concept of a compensated haemolytic state is clinically vital because it explains why many patients with significant haemolytic disorders do not present with anaemia.
Consider hereditary spherocytosis: RBC lifespan may be 20–40 days (vs normal 120 days). Yet the patient may have a haemoglobin of 11–13 g/dL because the marrow is producing 3–5 times the normal red cell output. The patient is well, but has:
• Mild jaundice (scleral icterus, often unnoticed)
• Mild splenomegaly
• Reticulocyte count 5–10% (chronically elevated)
• Pigment gallstones by age 30–40
This state is vulnerable to acute decompensation by three main mechanisms:
1. Haemolytic crisis: Acute acceleration of haemolysis (e.g., oxidant stress, infection) → haemoglobin drops acutely.
2. Aplastic crisis (parvovirus B19): Transient cessation of erythropoiesis → haemoglobin crashes over 1–2 weeks. Most dramatic in sickle-cell disease and hereditary spherocytosis.
3. Megaloblastic crisis: Chronic high-turnover erythropoiesis → folate depletion → megaloblastic change → superimposed ineffective erythropoiesis → haemoglobin falls.
Recognising the compensated state — jaundice + splenomegaly + elevated reticulocytes but no or mild anaemia — is an important clinical pattern that prevents under-diagnosis.
SELF-CHECK
Which combination of laboratory findings most specifically points to intravascular rather than extravascular haemolysis?
A. ↑Reticulocyte count + ↑unconjugated bilirubin + splenomegaly
B. ↓Haptoglobin + haemoglobinuria + urinary haemosiderin
C. ↑LDH + ↑unconjugated bilirubin + polychromasia on smear
D. Spherocytes on smear + positive direct Coombs test
Reveal Answer
Answer: B. ↓Haptoglobin + haemoglobinuria + urinary haemosiderin
Markedly ↓haptoglobin (from free Hb binding and clearance), haemoglobinuria (free Hb crossing the renal threshold), and urinary haemosiderin (iron accumulation in renal tubular cells from chronic free Hb filtration) are the triad specific to intravascular haemolysis. Options A and C represent general markers of haemolysis shared by both types. Option D (spherocytes + positive DAT) points to warm AIHA — predominantly extravascular haemolysis.
Looking Ahead — SDL2 and SDL3 Categories

Looking Ahead: Hereditary and Acquired Haemolytic Anaemias
The classification frameworks and laboratory markers established in this SDL apply to all haemolytic anaemias. The next two SDLs in this cluster zoom into specific categories:
SDL2 — Hereditary Haemolytic Anaemias (PA16.2–PA16.5)
• Membrane defects: Hereditary spherocytosis (spectrin/ankyrin/band 3 mutations) — most common hereditary haemolytic anaemia in North Indians; hereditary elliptocytosis.
• Enzyme defects: G6PD deficiency (X-linked, most common enzyme deficiency worldwide; especially prevalent in Indians); pyruvate kinase deficiency.
• Haemoglobin defects: Sickle-cell disease (HbS/HbS) — vasoocclusion + haemolysis; the thalassaemia syndromes (alpha and beta) — microcytic anaemia + variable haemolysis.
SDL3 — Acquired Haemolytic Anaemias (PA16.6–PA16.8)
• Immune haemolytic anaemias: Warm AIHA (IgG, 37°C, extravascular), cold AIHA / cold agglutinin disease (IgM, <4°C, intravascular/agglutination), drug-induced (three mechanisms).
• Non-immune (microangiopathic) haemolytic anaemias (MAHA): DIC, TTP, HUS, pre-eclampsia/HELLP — schistocytes are the smear hallmark.
• Other acquired causes: PNH (GPI-anchor defect, CD55/CD59 loss), malaria, hypersplenism, mechanical destruction (prosthetic valves).
As you move through SDL2 and SDL3, revisit this SDL's classification tables — every specific disorder fits neatly into the axes you have just learned.