Page 1 of 16
AS8.1-2 | Pain Physiology and Clinical Pain Assessment — SDL Guide
CLINICAL SCENARIO
A 68-year-old woman with metastatic breast cancer is admitted to the oncology ward. The nursing team documents her pain as '6 out of 10' on the numeric rating scale, but she has barely eaten, sleeps only in short fragmented bursts, and winces when repositioned. Her son insists the morphine dose 'should be enough by now.' Her attending anaesthesiologist, called in consultation, begins not by adjusting the opioid but by asking a precise set of questions — onset, site, character, radiation, aggravating factors — and then by watching her face during movement. Within thirty minutes, a previously unrecognised neuropathic component is identified and a targeted adjuvant is added. Pain management done well begins not with prescribing, but with understanding: understanding the anatomy of nociception, the physiological journey from tissue injury to conscious perception, and the disciplined clinical art of translating a patient's lived suffering into actionable clinical data.
WHY THIS MATTERS
Pain is the most common reason patients seek medical attention worldwide, and it is simultaneously the most undertreated condition in hospitalised patients. For an anaesthesiologist, a granular understanding of pain physiology is not optional background knowledge — it is the mechanistic foundation upon which every analgesic intervention is rationally designed. Why does a local anaesthetic nerve block abolish somatic pain but leave visceral pain intact? Why does wind-up in the dorsal horn explain why a patient's pain increases over hours even after the precipitating injury has stabilised? Why does neuropathic pain respond poorly to paracetamol but sometimes dramatically to low-dose amitriptyline? These questions cannot be answered without understanding peripheral sensitisation, central sensitisation, descending modulation, and the anatomy of spinothalamic projections. Equally, competent clinical pain assessment — using validated tools, adjusting for the patient's age, cognition, language, and clinical context — is the skill that makes pharmacological and interventional precision possible. The NMC 2024 competencies AS8.1 and AS8.2 demand that every final-year medical student reaches this standard before graduation.
RECALL
Before proceeding, refresh your recall of three foundational domains. First, from neuroanatomy (AN): the dorsal root ganglion houses the cell bodies of primary afferent neurons; the spinothalamic tract carries pain and temperature from the contralateral spinal cord to the thalamus and cortex. Second, from physiology (PY): action potential generation requires a threshold change in membrane potential driven by sodium influx through voltage-gated Na⁺ channels; summation — both temporal and spatial — amplifies subthreshold inputs. Third, from pharmacology (PH/BI): prostaglandins are synthesised from arachidonic acid via cyclooxygenase (COX-1 and COX-2); non-steroidal anti-inflammatory drugs (NSAIDs) inhibit these enzymes and thereby reduce peripheral sensitisation of nociceptors. These recall points form the scaffold onto which today's content is built.
Nociceptors: Types, Distribution, and Peripheral Sensitisation
Nociceptors are high-threshold, free nerve endings of primary afferent neurons that respond specifically to potentially tissue-damaging stimuli. They are not passive sensors; they are dynamic transducers whose sensitivity is tightly regulated by the local chemical environment. Understanding their biology is the first step in understanding why pain so often outlasts the injury that triggered it.
Primary afferent fibre types are classified by diameter and myelination. Aδ (A-delta) fibres are thinly myelinated, 2–5 μm in diameter, and conduct at 5–30 m/s. They mediate the initial sharp, well-localised 'first pain' — the sensation experienced immediately when you touch a hot surface. C fibres are unmyelinated, 0.2–1.5 μm in diameter, and conduct at 0.5–2 m/s. They are responsible for the slower, diffuse, burning, aching 'second pain' that follows. C fibres account for approximately 80% of all nociceptive afferents, making them the dominant vehicle for clinical pain transmission.
Nociceptor transduction is mediated by a family of ion channels that convert mechanical, thermal, or chemical energy into depolarising currents. TRPV1 (transient receptor potential vanilloid 1) is activated by temperatures above 43 °C, acidic pH, and capsaicin — the compound responsible for chilli pepper heat. TRPA1 responds to cold, mechanical deformation, and reactive electrophilic compounds. PIEZO2 channels mediate mechanotransduction. Inflammatory mediators released following tissue injury — including prostaglandin E₂ (PGE₂), bradykinin, serotonin, histamine, substance P, and nerve growth factor (NGF) — lower the activation thresholds of TRPV1 and other channels, a process termed peripheral sensitisation.
Peripheral sensitisation produces two clinically important phenomena: primary hyperalgesia, defined as increased pain sensitivity at the site of injury (a burn feels more painful when touched gently than intact skin), and allodynia at the wound margin, where normally innocuous stimuli such as light touch become painful. The analgesic consequences are profound: NSAIDs and corticosteroids reduce peripheral sensitisation by limiting prostaglandin synthesis and inflammatory signalling, which is why they are most effective when given before or early after an injurious stimulus.
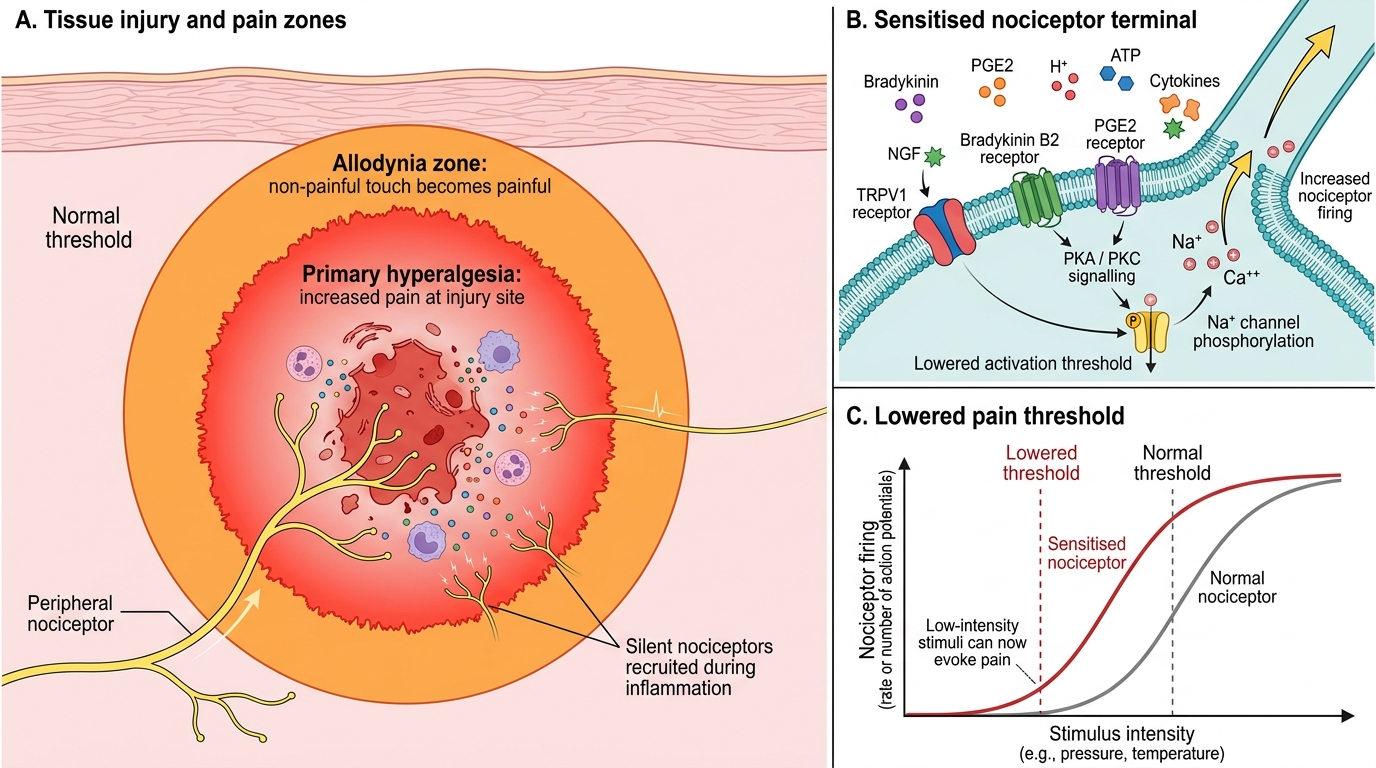
Peripheral Sensitisation After Tissue Injury
A clinically important concept is the nociceptor 'sleeping' population: approximately 30% of nociceptors are silent under normal physiological conditions and are recruited only in the context of significant inflammation. This explains why an inflamed joint is exquisitely tender, whereas the same mechanical load applied to the contralateral healthy joint causes no discomfort.
Spinal Cord Processing: Dorsal Horn, Wind-Up, and Central Sensitisation
Central sensitisation is the phenomenon by which the central nervous system amplifies nociceptive signals, producing pain responses that are disproportionate to — or independent of — peripheral nociceptive input. It is the neurophysiological substrate of many chronic pain syndromes and is the reason why analgesic strategies targeted exclusively at the periphery are often insufficient.
Primary afferent fibres synapse on neurons in the dorsal horn of the spinal cord, specifically in Rexed laminae I, II (substantia gelatinosa), and V. Aδ fibres project predominantly to laminae I and V; C fibres to laminae I and II. The primary neurotransmitters released from primary afferents are glutamate and substance P. Glutamate acts on three receptor types: AMPA receptors (fast, sodium-mediated, producing immediate excitation), kainate receptors, and NMDA (N-methyl-D-aspartate) receptors. Under normal conditions, the NMDA receptor pore is blocked by a magnesium ion at resting membrane potential — a voltage-dependent block that prevents its activation by single stimuli.
Wind-up occurs when C fibres fire repeatedly at frequencies above approximately 0.5 Hz. Repetitive stimulation causes progressive depolarisation of the dorsal horn neuron. When the membrane potential rises sufficiently, the Mg²⁺ block is relieved, and NMDA receptors become activated. This leads to large calcium influx, activation of intracellular kinases (PKC, ERK), and transcription of pro-excitatory genes — a process that dramatically increases the neuron's responsiveness to subsequent stimuli. The clinical correlate of wind-up is the observation that a patient's pain escalates over the first hours or days after surgery or acute injury even as tissue healing progresses.
Central sensitisation encompasses persistent wind-up, expanded receptive fields (so pain spreads to uninjured areas — termed secondary hyperalgesia), and altered response properties. Ketamine, an NMDA receptor antagonist, is effective specifically because it interrupts this positive-feedback loop. Sub-anaesthetic doses of ketamine administered perioperatively reduce wind-up and therefore the total analgesic requirement — the mechanistic basis of its use in multimodal analgesia.
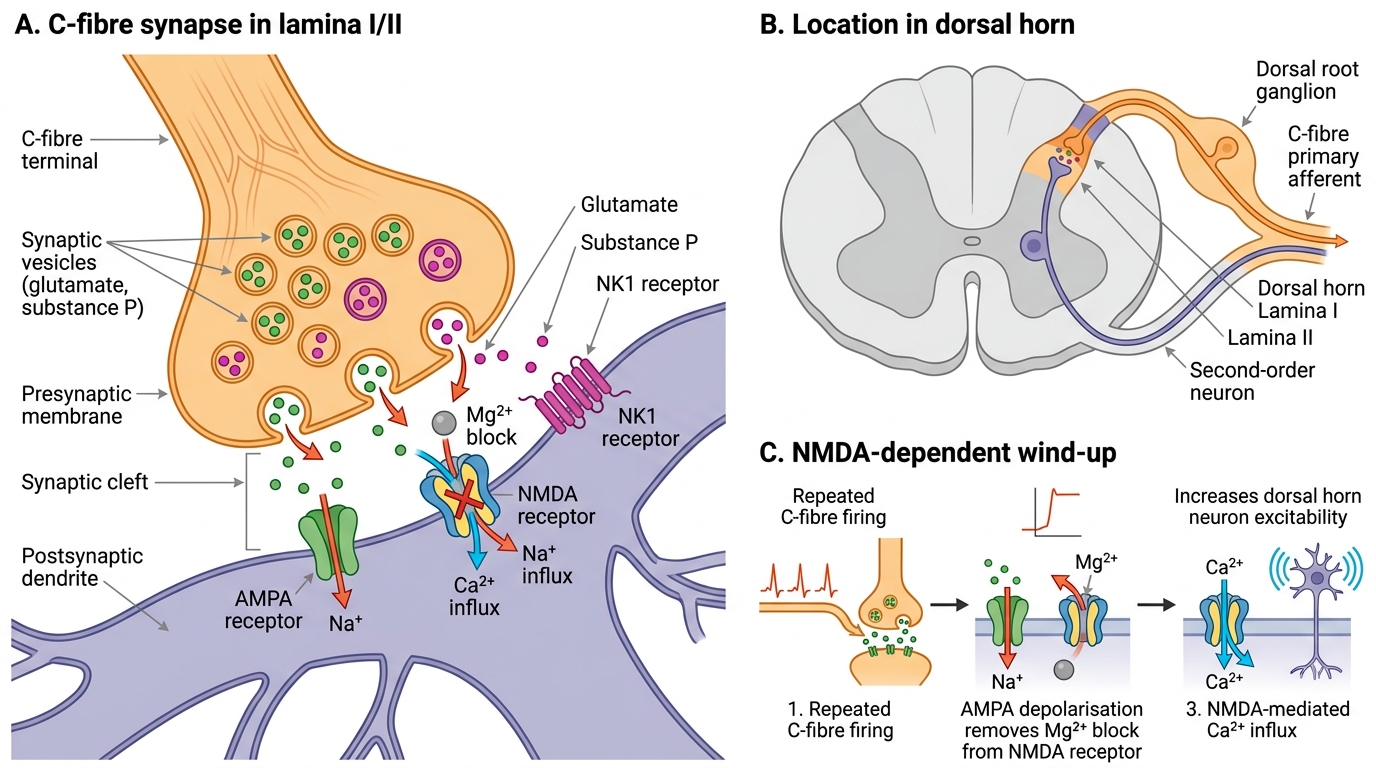
Dorsal Horn C-Fibre Synapse and NMDA-Mediated Wind-Up
Descending modulation — the bidirectional influence of brainstem and cortical centres on dorsal horn excitability — is equally important. The periaqueductal grey (PAG) and rostroventromedial medulla (RVM) project to the dorsal horn via serotonergic and noradrenergic pathways. These descending systems can be either inhibitory (reducing pain) or facilitatory (amplifying pain). Endogenous opioids (enkephalins, endorphins) act at μ, δ, and κ opioid receptors in both the PAG and the dorsal horn, producing analgesia — the mechanism co-opted by exogenous opioid drugs. Descending noradrenergic inhibition accounts for the analgesic efficacy of tricyclic antidepressants and serotonin-norepinephrine reuptake inhibitors (SNRIs) such as duloxetine and venlafaxine in neuropathic pain states.
Ascending Pathways and Cortical Pain Processing
From the dorsal horn, second-order neurons decussate in the anterior white commissure and ascend in the anterolateral system, principally via the spinothalamic tract (STT). The STT projects to the ventroposterolateral (VPL) nucleus of the thalamus, which in turn relays signals to the primary somatosensory cortex (S1) for precise localisation and intensity coding. A parallel projection to the medial thalamic nuclei reaches the anterior cingulate cortex (ACC) and insular cortex, which encode the affective-motivational dimension of pain — the suffering and unpleasantness that gives pain its urgency and that persists even when intensity diminishes.
This anatomical duality explains why two patients with identical tissue injuries can report radically different pain experiences: the sensory-discriminative dimension (location, quality, intensity) is broadly consistent, but the affective-motivational dimension (distress, fear, disability) varies enormously with psychological state, prior experience, cultural background, and the presence of anxiety or depression. Pain catastrophising — the tendency to ruminate on pain and feel helpless — amplifies activity in the ACC and correlates with worse analgesic outcomes, explaining why psychological interventions are an evidence-based component of chronic pain management.
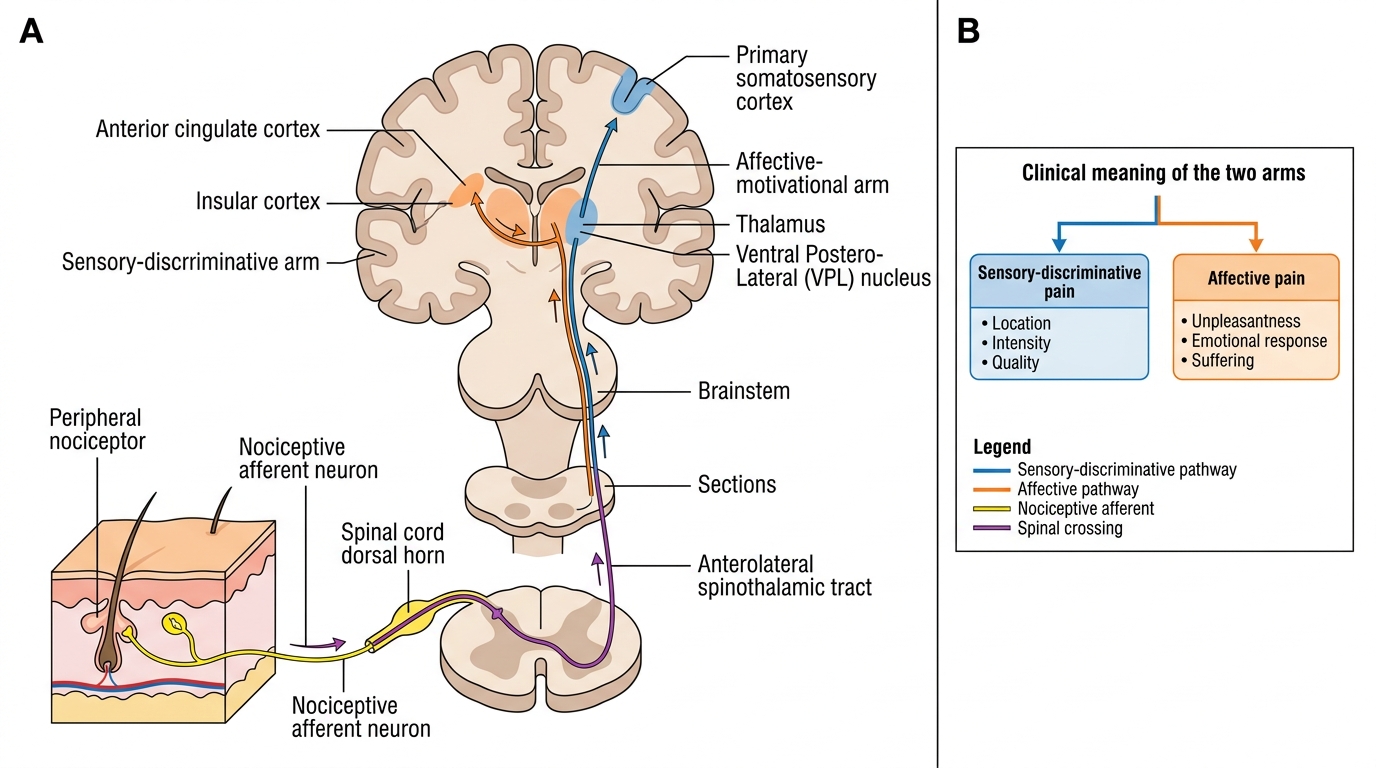
Ascending Pain Pathway
The distinction between nociceptive pain and neuropathic pain is clinically and mechanistically fundamental. Nociceptive pain — somatic or visceral — results from activation of an intact nociceptive system by a noxious stimulus and resolves when the stimulus is removed or the tissue heals. Neuropathic pain results from injury or disease of the somatosensory nervous system itself (peripheral or central). Its hallmarks are: burning, electric-shock, or shooting quality; allodynia; hyperalgesia extending beyond the zone of injury; and spontaneous pain in the absence of ongoing stimulus. Examples include diabetic peripheral neuropathy, postherpetic neuralgia, phantom limb pain, and central post-stroke pain. Neuropathic pain responds poorly to NSAIDs and requires adjuvants such as gabapentinoids (gabapentin, pregabalin), tricyclic antidepressants, SNRIs, or topical lidocaine patches. Identifying the neuropathic component of a patient's pain is therefore a critical clinical assessment skill.