Page 2 of 7
BI4.1-8 | Chemistry and Metabolism of Lipids — SDL Guide (Part 2)
Cholesterol — Synthesis, Transport, and Regulation (BI4.5)
Cholesterol is a 27-carbon sterol — a rigid, planar molecule with a characteristic four-ring steroid nucleus (3 cyclohexane + 1 cyclopentane) and a hydroxyl group at C-3. Despite its reputation as a 'bad' molecule, cholesterol is essential for life:

Figure: Cholesterol — Synthesis, Transport, and Regulation (BI4.5)
- Cell membranes — cholesterol inserts between phospholipids, modulating membrane fluidity
- Bile acids — synthesised from cholesterol in the liver, essential for fat digestion
- Steroid hormones — cortisol, aldosterone, testosterone, oestrogen, progesterone
- Vitamin D — synthesised from 7-dehydrocholesterol in skin on UV exposure
Cholesterol synthesis (de novo):
Occurs in the cytoplasm and smooth endoplasmic reticulum of virtually all nucleated cells, but primarily the liver.
The pathway: Acetyl-CoA → Acetoacetyl-CoA → HMG-CoA → Mevalonate → → → Squalene → Lanosterol → Cholesterol
The rate-limiting enzyme is HMG-CoA reductase — the enzyme that converts HMG-CoA to mevalonate. This is the target of statins (the most prescribed drug class in the world). Atorvastatin, rosuvastatin, and simvastatin are competitive inhibitors of HMG-CoA reductase → they block cholesterol synthesis in the liver → the liver upregulates LDL receptors to pull cholesterol from the blood → blood LDL levels fall.
Regulation of HMG-CoA reductase:
• SREBP (sterol regulatory element-binding protein) — when intracellular cholesterol is LOW, SREBP is released from the ER membrane, travels to the nucleus, and activates transcription of HMG-CoA reductase and LDL receptor genes. When cholesterol is HIGH, SREBP stays sequestered in the ER membrane → transcription stops.
• Hormonal: Insulin and thyroid hormones upregulate; glucagon downregulates
• Feedback inhibition: Cholesterol and its oxysterol derivatives inhibit HMG-CoA reductase directly
Cholesterol excretion:
The body cannot break the steroid ring → cholesterol cannot be degraded to CO₂ and water. The only significant route of excretion is through bile acids and free cholesterol in bile. Bile acids are recycled via the enterohepatic circulation (liver → bile → intestine → reabsorbed in ileum → portal vein → back to liver). Bile acid sequestrants (cholestyramine) interrupt this cycle → more cholesterol is diverted to bile acid synthesis → blood cholesterol falls.
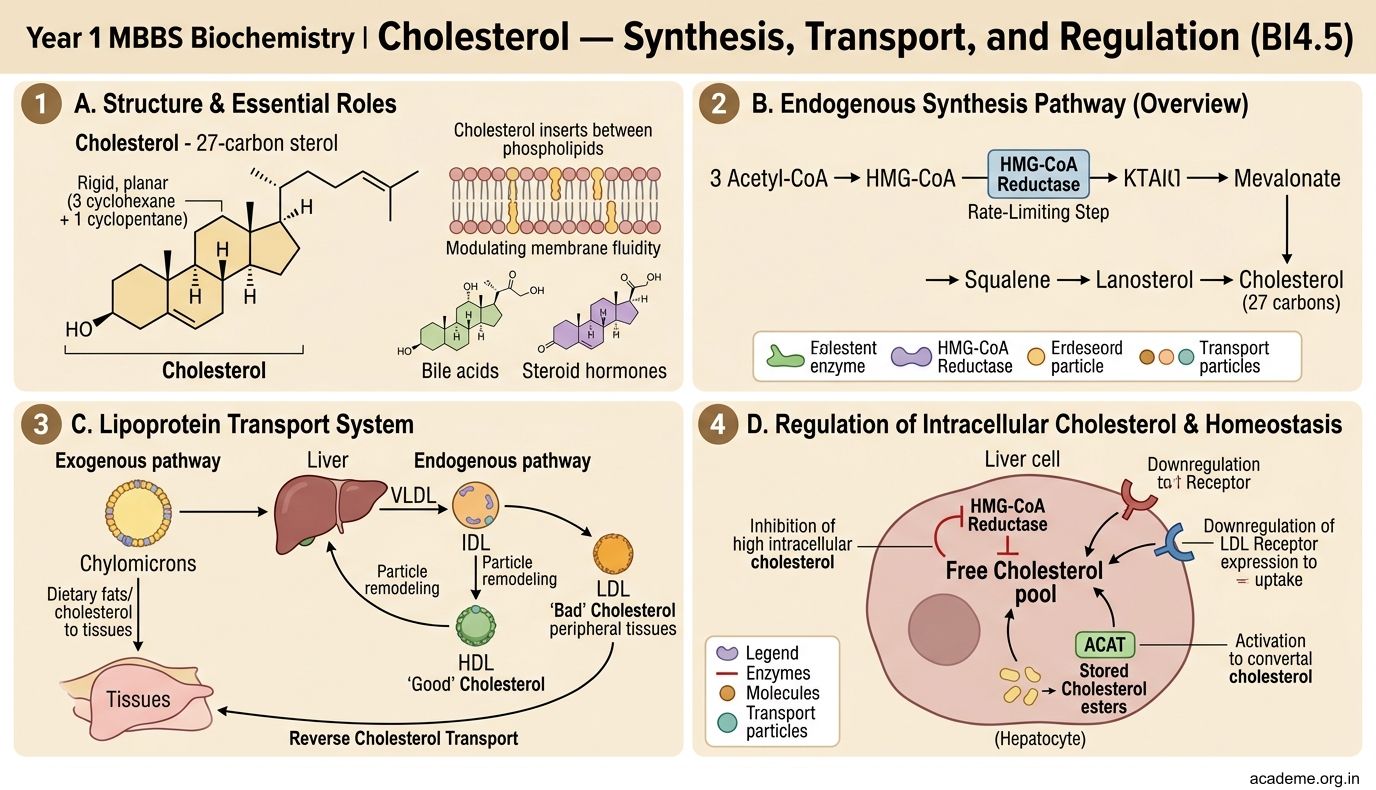
Phospholipids and Glycolipids — Membranes and Myelin (BI4.6)
Phospholipids are the architectural backbone of every cell membrane. They have a glycerol backbone with two fatty acids (hydrophobic tails) and a phosphate group linked to a polar head group (hydrophilic head). This amphipathic structure (one end water-loving, one end water-fearing) is what allows them to spontaneously form lipid bilayers — the basis of all biological membranes.

Figure: Phospholipids and Glycolipids — Membranes and Myelin (BI4.6)
Major glycerophospholipids:
• Phosphatidylcholine (lecithin) — the most abundant phospholipid in cell membranes. Also the major component of lung surfactant (dipalmitoylphosphatidylcholine, DPPC). Premature babies lacking surfactant develop respiratory distress syndrome (RDS) — the L/S ratio (lecithin/sphingomyelin ratio > 2) in amniotic fluid indicates lung maturity.
• Phosphatidylethanolamine — second most abundant membrane phospholipid
• Phosphatidylserine — normally on the inner leaflet of the cell membrane; its appearance on the outer leaflet signals apoptosis (programmed cell death) — macrophages recognise this 'eat me' signal
• Phosphatidylinositol — precursor to IP₃ and DAG (second messengers in cell signalling)
• Cardiolipin — found exclusively in the inner mitochondrial membrane, essential for electron transport chain function
Sphingolipids — based on the amino alcohol sphingosine (not glycerol):
• Sphingomyelin — abundant in the myelin sheath of nerves. Degraded by sphingomyelinase — deficiency causes Niemann-Pick disease (sphingomyelin accumulates in brain, liver, spleen → neurodegeneration, hepatosplenomegaly).
Glycolipids — contain one or more sugar residues:
• Cerebrosides — sphingosine + fatty acid + one sugar (galactose in brain, glucose elsewhere). Degraded by galactocerebrosidase — deficiency causes Krabbe disease (leukodystrophy).
• Gangliosides — contain oligosaccharides with sialic acid (NANA). Abundant in neuronal membranes. GM₂ ganglioside accumulates in Tay-Sachs disease (hexosaminidase A deficiency) — progressive neurodegeneration in infants, cherry-red spot on macula, death by age 3-4.
• Sulfatides — sulfated cerebrosides. Accumulate in metachromatic leukodystrophy (arylsulfatase A deficiency).
Clinical pattern: The lipid storage diseases (sphingolipidoses) are all caused by deficiency of lysosomal enzymes that degrade specific sphingolipids. Each missing enzyme → specific lipid accumulates → specific organ damage. Tay-Sachs, Niemann-Pick, Gaucher disease, Krabbe disease, and Fabry disease all follow this pattern.
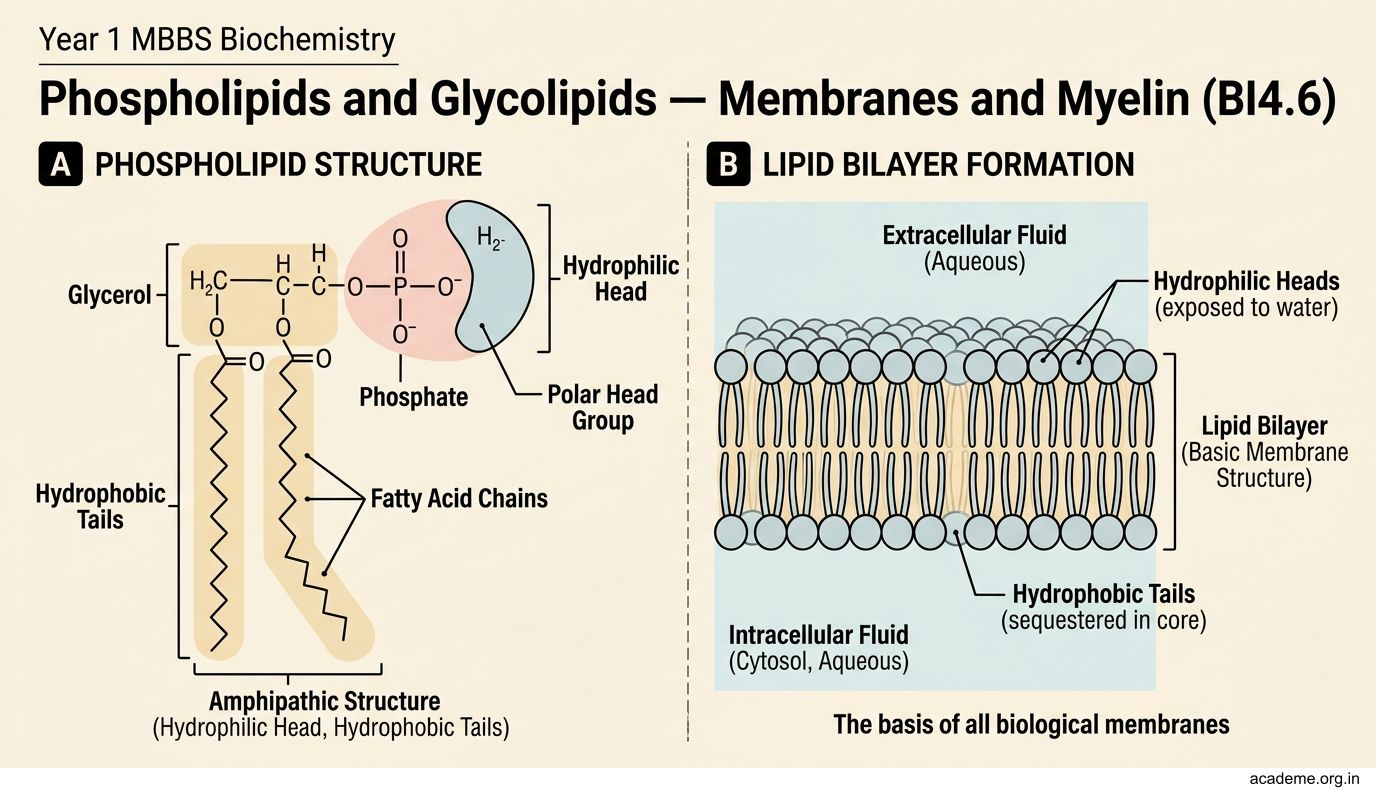
Lipoproteins — How Fat Travels in Blood (BI4.7)
Lipids are insoluble in water — so how do they travel in the aqueous environment of blood? The answer is lipoproteins — spherical particles with a hydrophobic core (triglycerides and cholesterol esters) surrounded by a hydrophilic shell (phospholipids, free cholesterol, and apolipoproteins).

Figure: Lipoproteins — How Fat Travels in Blood (BI4.7)
There are five major classes, arranged by density (lowest to highest):
1. Chylomicrons — the largest and least dense lipoproteins
• Origin: intestinal epithelial cells (enterocytes)
• Function: transport dietary (exogenous) triglycerides from gut to tissues
• Key apolipoprotein: ApoB-48 (identifies it as intestinal), ApoCII (activates lipoprotein lipase), ApoE (liver uptake)
• Pathway: Dietary fat → chylomicrons → lymphatics (thoracic duct) → blood → lipoprotein lipase (LPL) on capillary endothelium cleaves triglycerides → fatty acids taken up by muscle/adipose → chylomicron remnants → taken up by liver (via ApoE receptor)
• Fasting serum should have NO chylomicrons — their presence indicates a recent meal or a metabolic defect
2. VLDL (Very Low-Density Lipoprotein)
• Origin: liver
• Function: transport endogenous triglycerides (made in the liver) to tissues
• Key apolipoprotein: ApoB-100, ApoCII, ApoE
• Pathway: VLDL → LPL removes triglycerides → IDL (intermediate-density lipoprotein) → hepatic lipase removes more triglycerides → LDL
3. LDL (Low-Density Lipoprotein) — the 'bad cholesterol'
• Origin: product of VLDL metabolism (VLDL → IDL → LDL)
• Function: delivers cholesterol to peripheral tissues
• Key apolipoprotein: ApoB-100 (one molecule per LDL particle — the ligand for the LDL receptor)
• Pathway: LDL binds to LDL receptors on cells → receptor-mediated endocytosis → cholesterol released inside the cell for membranes and steroid synthesis → LDL receptor recycled
• When LDL is elevated or LDL receptors are deficient, LDL accumulates in blood → penetrates the arterial wall → oxidised LDL → taken up by macrophages → FOAM CELLS → atherosclerotic plaque.
4. HDL (High-Density Lipoprotein) — the 'good cholesterol'
• Origin: liver and intestine (secreted as nascent/disc-shaped HDL)
• Function: reverse cholesterol transport — picks up excess cholesterol from peripheral tissues and returns it to the liver for excretion as bile acids
• Key apolipoprotein: ApoA-I (activates LCAT — lecithin-cholesterol acyltransferase, which esterifies cholesterol on HDL surface → cholesterol ester moves to core → HDL matures from disc to sphere)
• HDL also transfers cholesterol esters to VLDL/LDL via CETP (cholesteryl ester transfer protein) in exchange for triglycerides
• High HDL is PROTECTIVE — it removes cholesterol from arteries. Low HDL is an independent risk factor for cardiovascular disease.
5. Lipoprotein(a) [Lp(a)] — a modified LDL particle with an additional apo(a) protein. Lp(a) levels are genetically determined and NOT lowered by diet or statins. Elevated Lp(a) is an independent risk factor for cardiovascular disease and is particularly prevalent in the Indian population.
Atherosclerosis and Dyslipidaemia — When Lipids Kill (BI4.8)
Atherosclerosis is the pathological process by which cholesterol-laden plaques build up in arterial walls. It is the biochemical basis of coronary artery disease, stroke, and peripheral vascular disease — collectively the leading cause of death worldwide.

Figure: Dyslipidaemias — the biochemical disorders:

Figure: The mechanism (step by step):

Figure: Atherosclerosis and Dyslipidaemia — When Lipids Kill (BI4.8)
The mechanism (step by step):
- Endothelial injury — caused by hypertension, smoking, diabetes, or turbulent blood flow at arterial branch points. The damaged endothelium becomes 'leaky'.
- LDL infiltration — LDL particles enter the subendothelial space (intima) of the arterial wall.
- Oxidation — trapped LDL undergoes oxidative modification (by reactive oxygen species). Oxidised LDL (oxLDL) is not recognised by normal LDL receptors.
- Foam cell formation — macrophages express scavenger receptors (SR-A, CD36) that take up oxLDL without limit (unlike LDL receptors, scavenger receptors are NOT downregulated by intracellular cholesterol). Macrophages gorge on oxLDL → become bloated foam cells → the earliest visible lesion: the fatty streak.
- Plaque progression — foam cells release inflammatory cytokines → recruit more macrophages and smooth muscle cells → smooth muscle cells migrate from the media into the intima and produce a fibrous cap over the growing lipid core → the atherosclerotic plaque.
- Plaque rupture — if the fibrous cap is thin (due to matrix metalloproteinases from macrophages), it can rupture → the thrombogenic lipid core is exposed to blood → platelet aggregation and thrombus formation → acute occlusion → myocardial infarction or stroke.
Cross-reference to Anatomy (AN22.3-AN22.4): The coronary arteries you studied in Anatomy — LAD, LCx, RCA — are where this process occurs. LAD occlusion causes anterior MI (the 'widow-maker'). RCA occlusion causes inferior MI. The anatomy of which artery feeds which territory determines what dies when a plaque ruptures.
Dyslipidaemias — the biochemical disorders:
- Type IIa (Familial Hypercholesterolaemia, FH) — defective or absent LDL receptors → LDL cannot be cleared from blood → severe hypercholesterolaemia. Heterozygous FH (1 in 250) → LDL 300-500 mg/dL → MI by age 40-50. Homozygous FH (1 in 1,000,000) → LDL >600 mg/dL → MI in childhood. FH is the most common inherited metabolic disease.
- Type IV (Familial Hypertriglyceridaemia) — increased VLDL production → high triglycerides. Risk: acute pancreatitis when TG >1000 mg/dL.
- Type I (Familial Hyperchylomicronaemia) — deficiency of lipoprotein lipase or ApoCII → chylomicrons cannot be cleared → milky serum (lipaemia), eruptive xanthomas, pancreatitis.
Lipid-lowering drugs — mechanism of action:
| Drug Class | Mechanism | Primary Effect |
|---|---|---|
| Statins (atorvastatin, rosuvastatin) | Inhibit HMG-CoA reductase → ↓ liver cholesterol → ↑ LDL receptors → ↓ blood LDL | ↓ LDL 30-50% |
| Fibrates (fenofibrate, gemfibrozil) | Activate PPARα → ↑ LPL activity → ↑ fatty acid oxidation | ↓ Triglycerides 40-60% |
| Ezetimibe | Blocks NPC1L1 transporter in intestinal brush border → ↓ cholesterol absorption | ↓ LDL 15-20% |
| PCSK9 inhibitors (evolocumab) | Block PCSK9 → LDL receptors are not degraded → more LDL receptors on liver → ↓ blood LDL | ↓ LDL 50-70% |
| Bile acid sequestrants (cholestyramine) | Bind bile acids in gut → interrupt enterohepatic circulation → liver diverts cholesterol to bile acid synthesis | ↓ LDL 15-30% |
| Omega-3 fatty acids (EPA/DHA) | ↓ VLDL synthesis in liver, ↓ triglyceride synthesis | ↓ Triglycerides 20-30% |
SELF-CHECK
A 45-year-old man has a total cholesterol of 380 mg/dL and LDL of 310 mg/dL despite a healthy diet. His father died of a heart attack at age 42. Physical examination reveals tendon xanthomas on the Achilles tendons. What is the most likely diagnosis, and which receptor is defective?
A. Familial hypertriglyceridaemia; lipoprotein lipase receptor
B. Familial hypercholesterolaemia; LDL receptor defect (heterozygous)
C. Type I hyperlipoproteinaemia; ApoCII deficiency
D. Metabolic syndrome; insulin receptor dysfunction
Reveal Answer
Answer: B. Familial hypercholesterolaemia; LDL receptor defect (heterozygous)
This is heterozygous familial hypercholesterolaemia (FH) — the most common inherited metabolic disease (1 in 250). The LDL receptor is defective or reduced in number → LDL cannot be cleared from blood → markedly elevated LDL cholesterol → cholesterol deposits in tendons (xanthomas), arteries (premature atherosclerosis), and around the eyes (xanthelasma). Family history of premature MI is a hallmark. Treatment: high-dose statins + ezetimibe, and increasingly PCSK9 inhibitors.
CLINICAL PEARL
The Indian paradox in cardiovascular disease: Indians develop coronary artery disease a full decade earlier than Western populations, and at lower LDL levels. Why? Several biochemical factors are at play: (1) Higher Lp(a) levels — genetically determined, not lowered by statins, prevalent in South Asians. (2) Insulin resistance and the metabolic syndrome — central obesity even at 'normal' BMI (the 'thin-fat Indian' phenotype). (3) Low HDL — Indian diets high in refined carbohydrates and trans fats suppress HDL. (4) High triglycerides — driven by carbohydrate-heavy diets. (5) Smaller, denser LDL particles — more atherogenic than large buoyant LDL, even at the same LDL-C level. This is why the standard Western cutoff for 'normal' LDL (130 mg/dL) may be too lenient for Indians — Indian guidelines recommend an LDL target of <100 mg/dL for moderate-risk patients and <70 mg/dL for high-risk patients.