Page 3 of 8
BI5.1-9 | Chemistry & Metabolism of Proteins and Immunology — SDL Guide (Part 3)
Specialised Products from Amino Acids and Inborn Errors (BI5.7)
Amino acids are not just protein building blocks — they are precursors for some of the body's most important molecules. When the enzymes that process them are missing, the consequences are devastating.

Figure: Specialised Products from Amino Acids and Inborn Errors (BI5.7)
Glycine — the simplest amino acid (R-group is just H):
- Precursor for: haem (we'll cover this next), purines (DNA building blocks), creatine (muscle energy reserve), glutathione (antioxidant), bile salts (conjugation)
- Glycine is also an inhibitory neurotransmitter in the spinal cord
Phenylalanine and Tyrosine:
- Phenylalanine → tyrosine (by phenylalanine hydroxylase, requires BH4 cofactor)
- Tyrosine is the precursor for: catecholamines (dopamine → noradrenaline → adrenaline), thyroid hormones (T3, T4), melanin (skin/hair pigment)
- Phenylketonuria (PKU) — deficiency of phenylalanine hydroxylase → Phe accumulates → musty/mousy odour in urine, intellectual disability if untreated. Detected by Guthrie test (newborn screening). Treatment: low-Phe diet from birth + tyrosine supplementation.
- Alkaptonuria — deficiency of homogentisic acid oxidase (in tyrosine degradation) → homogentisic acid accumulates → urine turns dark on standing (ochronosis), dark pigment deposits in cartilage. Relatively benign.
- Albinism — deficiency of tyrosinase → no melanin → white skin/hair, pink eyes, photosensitivity
Tryptophan:
- Precursor for: serotonin (mood neurotransmitter) → melatonin (sleep hormone), and NAD+/NADP+ (niacin — vitamin B3)
- Carcinoid syndrome — tryptophan is diverted to serotonin by the tumour → niacin deficiency → pellagra (dermatitis, diarrhoea, dementia)
- Hartnup disease — poor tryptophan absorption → pellagra-like symptoms
Methionine:
- Activated form is S-adenosylmethionine (SAM) — the body's universal methyl donor (methylation of DNA, RNA, proteins, phospholipids, neurotransmitters)
- Homocystinuria — deficiency of cystathionine beta-synthase → homocysteine accumulates → tall stature, lens subluxation (downward, unlike Marfan which is upward), intellectual disability, thromboembolism
Branched-chain amino acids (Val, Leu, Ile):
- Maple syrup urine disease (MSUD) — deficiency of branched-chain alpha-keto acid dehydrogenase → branched-chain amino acids and their keto acids accumulate → sweet-smelling urine, neurological damage, lethal if untreated
Newborn screening — most countries now screen for PKU, MSUD, homocystinuria, and other inborn errors using a heel-prick blood sample taken 48-72 hours after birth (tandem mass spectrometry). Early detection and dietary management prevent irreversible brain damage.
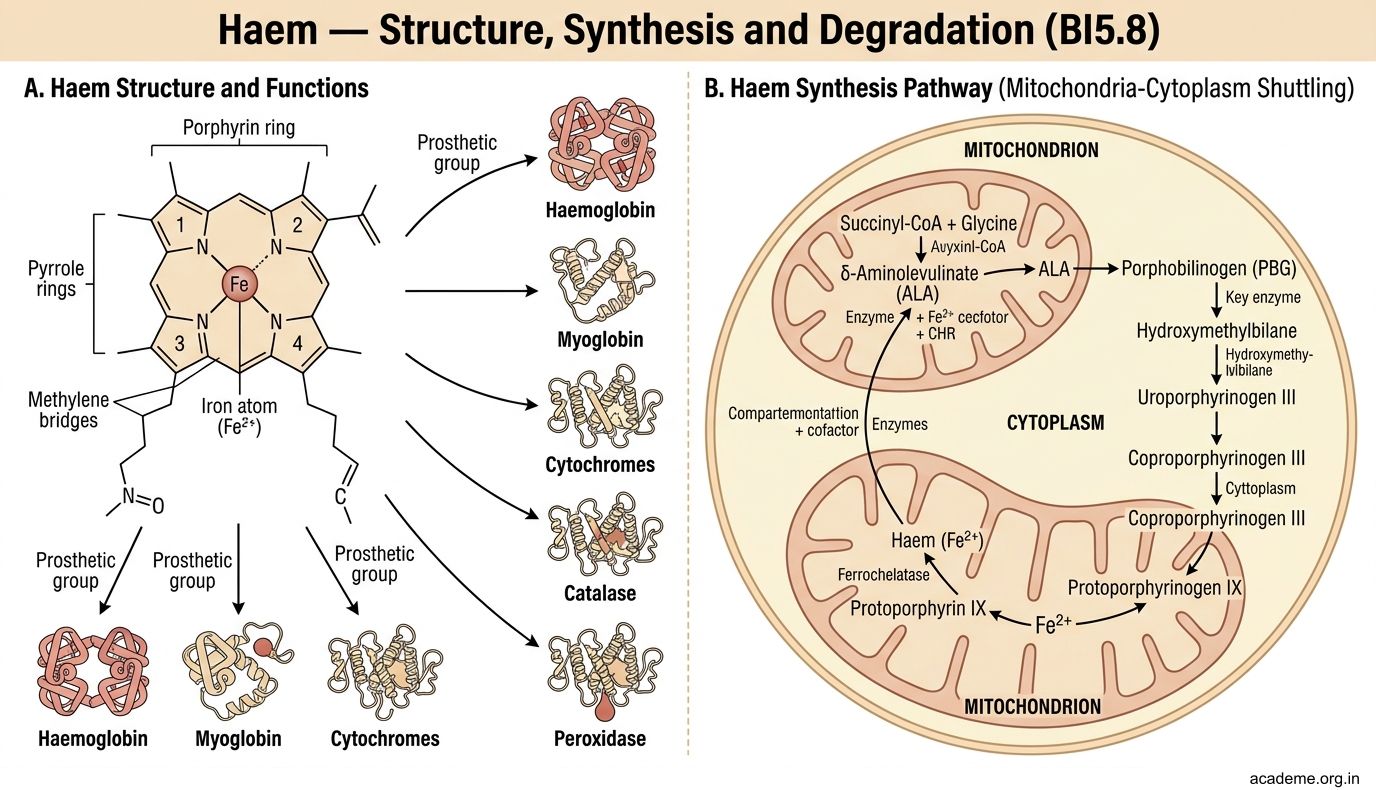
Haem — Structure, Synthesis and Degradation (BI5.8)
Haem is a flat, ring-shaped molecule made of four pyrrole rings joined together (porphyrin ring) with an iron atom (Fe2+) at the centre. It is the prosthetic group of haemoglobin, myoglobin, cytochromes, catalase, and peroxidase.

Figure: Haem — Structure, Synthesis and Degradation (BI5.8)
Haem synthesis (occurs in bone marrow and liver):
The pathway starts in the mitochondria, moves to the cytoplasm, and returns to the mitochondria:
- Succinyl-CoA + Glycine → delta-aminolevulinic acid (ALA) — catalysed by ALA synthase (rate-limiting step, mitochondria). Requires pyridoxal phosphate (vitamin B6) as cofactor. Inhibited by haem (feedback inhibition).
- 2 ALA → porphobilinogen (PBG) — catalysed by ALA dehydratase (cytoplasm). Inhibited by lead (this is why lead poisoning causes anaemia).
- PBG → ... → protoporphyrin IX (through several intermediates in cytoplasm)
- Protoporphyrin IX + Fe2+ → haem — catalysed by ferrochelatase (mitochondria). Also inhibited by lead.
Haem degradation (produces bilirubin → jaundice):
- Old RBCs are destroyed in the reticuloendothelial system (spleen, liver, bone marrow)
- Haem → biliverdin (green) → unconjugated bilirubin (yellow, lipid-soluble, toxic) — carried in blood bound to albumin
- Liver takes up unconjugated bilirubin → conjugated with glucuronic acid by UDP-glucuronyl transferase → conjugated bilirubin (water-soluble, non-toxic)
- Conjugated bilirubin is secreted into bile → reaches the intestine
- Gut bacteria convert it to urobilinogen → most is excreted as stercobilinogen/stercobilin (gives faeces their brown colour)
- Some urobilinogen is reabsorbed (enterohepatic circulation) → part is re-excreted by the liver, part goes to the kidney → urobilin (gives urine its yellow colour)
Types of jaundice:
- Pre-hepatic (haemolytic): excessive RBC destruction → unconjugated bilirubin rises → dark urine (urobilinogen), dark stools (excess stercobilin)
- Hepatic (hepatocellular): liver damage → both conjugated and unconjugated bilirubin rise
- Post-hepatic (obstructive): bile duct blocked → conjugated bilirubin rises → dark urine (conjugated bilirubin in urine), pale/clay-coloured stools (no stercobilin)
Neonatal jaundice — physiological jaundice in newborns occurs because the immature liver has low UDP-glucuronyl transferase activity. Usually resolves by day 10. If unconjugated bilirubin rises too high, it crosses the blood-brain barrier → kernicterus (bilirubin encephalopathy) → permanent brain damage. Treatment: phototherapy (blue light converts bilirubin to a water-soluble form that can be excreted without conjugation).
Porphyrias — inherited defects in haem synthesis enzymes → accumulation of porphyrin intermediates:
- Acute intermittent porphyria — defect in PBG deaminase → ALA and PBG accumulate → abdominal pain, neuropsychiatric symptoms, dark urine (porphyrin oxidation). Triggered by drugs (barbiturates, alcohol) that induce ALA synthase.
- Porphyria cutanea tarda — most common porphyria → photosensitive skin blisters (porphyrins absorb light and generate free radicals)
Haemoglobin — Types, Derivatives and Variants (BI5.9)
Haemoglobin (Hb) is the oxygen-carrying protein in red blood cells. Each molecule is a tetramer — 4 globin subunits, each carrying one haem group. So one Hb molecule carries up to 4 oxygen molecules.

Figure: Haemoglobin — Types, Derivatives and Variants (BI5.9)
Normal haemoglobin types:
- HbA (α2β2) — the major adult haemoglobin (~97% of adult Hb). Two alpha chains + two beta chains.
- HbA2 (α2δ2) — minor adult haemoglobin (~2-3%). Two alpha + two delta chains. Elevated in beta-thalassaemia trait (because beta chain production is reduced, delta chains compensate).
- HbF (α2γ2) — foetal haemoglobin (~60-80% at birth, falls to <1% by 6 months). Two alpha + two gamma chains. HbF has higher oxygen affinity than HbA — it snatches oxygen from maternal blood across the placenta. This is because HbF binds 2,3-bisphosphoglycerate (2,3-BPG) less effectively than HbA.
Haemoglobin derivatives:
- Oxyhaemoglobin (HbO2) — Hb bound to oxygen (bright red, in arterial blood)
- Deoxyhaemoglobin (deoxy-Hb) — Hb without oxygen (dark red/blue, in venous blood — this is why veins look blue through the skin)
- Carboxyhaemoglobin (HbCO) — Hb bound to carbon monoxide. CO has 250x greater affinity for Hb than O2 → even small amounts of CO displace O2 → tissue hypoxia. The patient looks cherry-red (not cyanotic), which is deceptive. Diagnosis: CO-oximetry (not pulse oximetry, which reads HbCO as HbO2). Treatment: 100% oxygen.
- Methaemoglobin (MetHb) — iron in haem is oxidised from Fe2+ to Fe3+ → cannot carry oxygen. Causes: nitrates/nitrites, dapsone, aniline dyes. Patients appear chocolate-brown cyanosis that does not respond to oxygen. Treatment: methylene blue IV (reduces Fe3+ back to Fe2+).
- Glycosylated haemoglobin (HbA1c) — glucose attaches non-enzymatically to the N-terminal valine of the beta chain. HbA1c reflects average blood glucose over 2-3 months (lifespan of RBC). Target in diabetes: <7%. Used for monitoring, NOT diagnosis in India (HbS and HbE interfere with some assays).
Haemoglobin variants (haemoglobinopathies):
- HbS (sickle haemoglobin) — point mutation: beta-6 Glu→Val. In deoxygenated state, HbS polymerises → sickle-shaped RBCs → vaso-occlusion (painful crises), haemolysis, organ damage. Heterozygotes (HbAS, sickle trait) are protected against falciparum malaria — this is why the sickle gene persists in malaria-endemic regions.
- HbC — beta-6 Glu→Lys. Milder than HbS. Target cells on blood film.
- HbE — beta-26 Glu→Lys. Common in Southeast Asia and northeast India. Usually mild. HbE/beta-thalassaemia is severe.
Thalassaemias — reduced production (not abnormal structure) of globin chains:
- Alpha-thalassaemia — reduced/absent alpha-chain synthesis. Four alpha genes (2 per chromosome 16). One deletion = silent carrier, two = alpha-thal trait, three = HbH disease (β4 tetramers), four = Hb Bart's (γ4 tetramers) → hydrops fetalis (fatal in utero).
- Beta-thalassaemia — reduced/absent beta-chain synthesis. Two beta genes (1 per chromosome 11). Beta-thal trait (minor) = one gene affected → mild microcytic anaemia, elevated HbA2. Beta-thal major (Cooley's anaemia) = both genes affected → severe anaemia from 6 months (when HbF should switch to HbA), transfusion-dependent, iron overload, bone marrow expansion (crew-cut skull on X-ray).
Spiral forward: In Physiology, you're learning about the oxygen dissociation curve — the sigmoid curve of HbA, left-shifted HbF, and how 2,3-BPG, pH, temperature, and CO2 shift the curve. The biochemistry you learn here (subunit structure, cooperativity, HbF affinity) is the molecular explanation for everything you see on that curve.
SELF-CHECK
A 3-month-old baby presents with severe anaemia requiring blood transfusion. Hb electrophoresis shows absent HbA, elevated HbF, and elevated HbA2. Which condition does this pattern suggest?
A. Sickle cell disease (HbSS)
B. Alpha-thalassaemia major (Hb Bart's)
C. Beta-thalassaemia major
D. Hereditary spherocytosis
Reveal Answer
Answer: C. Beta-thalassaemia major
Beta-thalassaemia major — both beta-globin genes are non-functional, so no HbA (α2β2) is produced. The baby survives on HbF (α2γ2) in utero, but as gamma-chain production naturally decreases after birth and beta chains can't replace them, severe anaemia develops around 3-6 months. HbA2 (α2δ2) is elevated because delta chains partially compensate. In sickle cell disease, you would see HbS, not absent HbA.