Page 9 of 18
FM10.{25-27,29} | Research Ethics & Ethics Committees — SDL Guide (Part 2)
Research Documentation and Ethical Research in Practice
The practical conduct of ethical research requires documentation systems that operationalise the principles laid down by IECs and the ICMR guidelines. These are the specific practices FM10.29 ('demonstrate ability to conduct research in pursuance of research ethics guidelines') requires students to internalise.
Essential documentation in a clinical trial or research project:
- Ethics approval letter — must be received before first participant enrolled; specifies which protocol version was approved.
- Informed consent form (ICF) — in the participant's language; signed before any research procedure; copies retained by participant and investigator.
- Case Report Form (CRF) — standardised data collection form; accuracy is a regulatory requirement.
- Investigator's Brochure (IB) — for Phase I–III drug trials; contains all available scientific information about the investigational product.
- Trial Master File (TMF) — archive of all essential documents that allows reconstruction of the trial's conduct.
- SAE reporting forms — serious adverse events must be reported to the IEC and CDSCO within specified timeframes (typically 24 hours for life-threatening events, 15 calendar days for other unexpected SAEs).
- Protocol deviations log — any unplanned departure from the approved protocol must be documented and reported to the IEC.
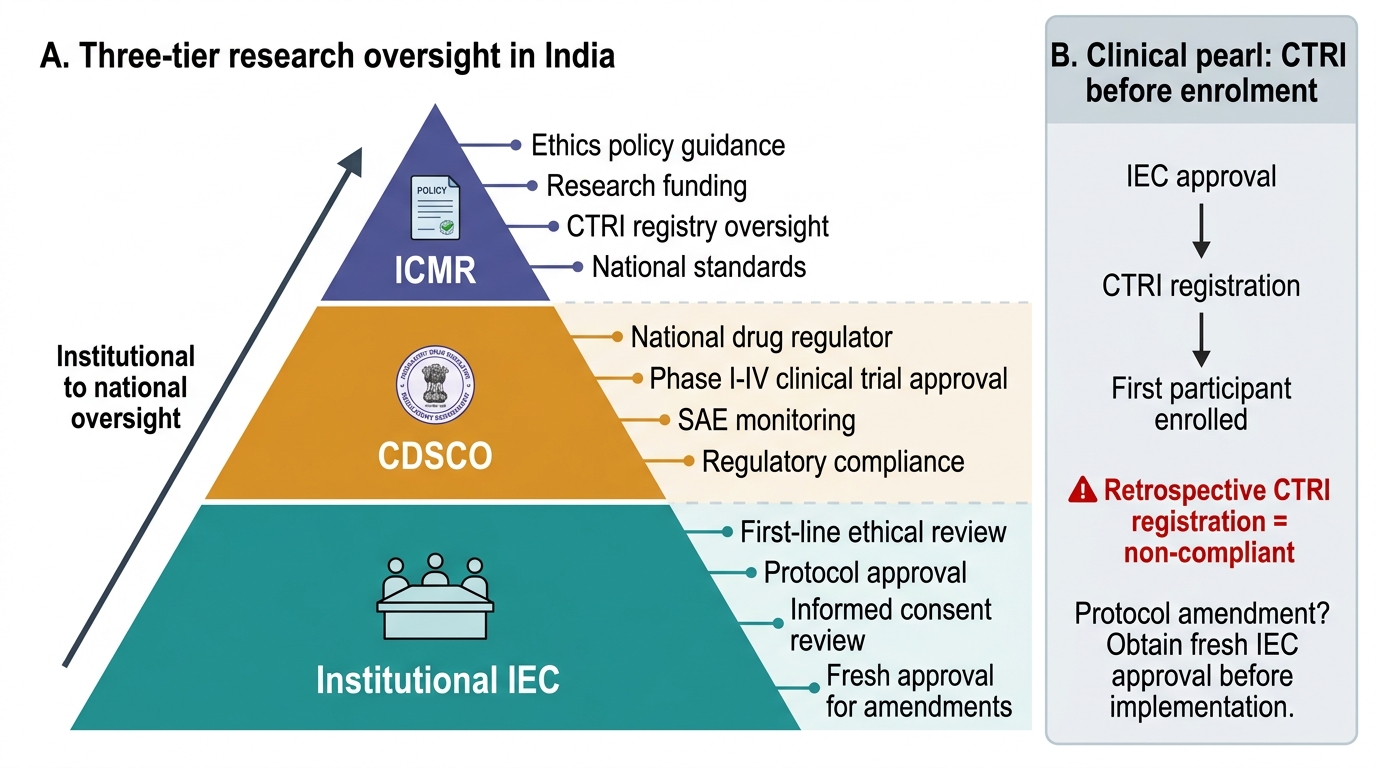
The three tiers of research oversight:
1. Institutional IEC — first-line review; no protocol may proceed without IEC approval.
2. CDSCO — national regulator; approves Phase I–IV drug and device trials; monitors SAE data.
3. ICMR — funds research and develops policy; maintains the Clinical Trials Registry – India (CTRI), where all clinical trials must be prospectively registered.
Vulnerable population protections — ICMR 2017 guidelines identify specific populations requiring additional safeguards: children (require guardian consent + child assent where possible), pregnant and breastfeeding women (research may only be conducted if it cannot be done on non-pregnant adults and the risk to the foetus is justifiable), persons with mental illness or cognitive impairment (capacity assessment + consent from legally authorised representative + person's own assent where possible), and prisoners (coercion risk — very restricted).
Three-tier Research Oversight in India
CLINICAL PEARL
CTRI registration is mandatory and must precede enrolment: Every clinical trial in India must be prospectively registered on the Clinical Trials Registry – India (CTRI) before the first participant is enrolled. Retrospective registration — registering after data collection — renders the trial non-compliant with ICMR guidelines and CDSCO regulations, and the findings may not be publishable in major journals (ICMJE mandate). Researchers must also obtain fresh IEC approval for every protocol amendment, not merely notify the committee. The most common compliance failure in Indian institutional research is retrospective CTRI registration or IEC amendment not sought before implementation of protocol changes.
Applied Practice: Ethical Review Scenarios
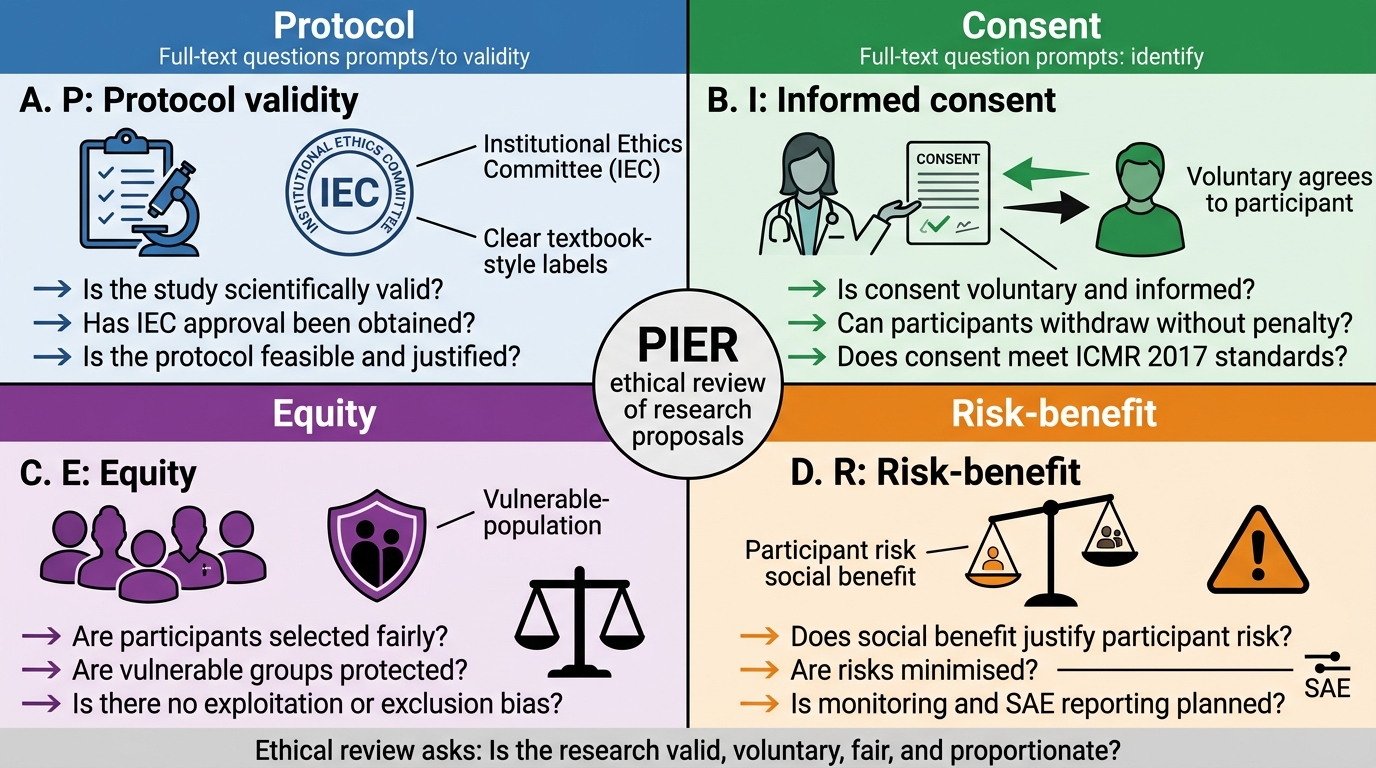
Applying research ethics principles to real scenarios is the competency FM10.29 specifically tests. The following framework — 'PIER' — is a structured approach to evaluating whether a research proposal or ongoing study is ethically permissible.
PIER Framework:
- P — Protocol validity: is the research question scientifically valid and answerable by the proposed methodology? Has the protocol been peer-reviewed and IEC-approved?
- I — Informed consent: has the consent process met all ICMR 2017 requirements — voluntary, informed, undocumented coercion absent, withdrawal without penalty, participant retains a copy?
- E — Equity: are participants selected fairly? Are vulnerable populations protected? Are the risks and benefits fairly distributed?
- R — Risk-benefit: do the potential social benefits of the research justify the risks imposed on participants? Are risks minimised to the extent possible?
Worked example 1 — the hook scenario: The pharmaceutical company's trial lacks IEC approval from the hospital's own IEC. Using a remote, unaffiliated IEC may not meet the 'institutional responsibility' principle of ICMR 2017 (each institution has responsibility for research conducted under its auspices). The per-patient enrollment fee is a potentially coercive financial inducement to the investigator (conflicts of interest). The resident should NOT enrol patients until (a) the hospital's own IEC reviews and approves, (b) the protocol is registered on CTRI, and (c) a proper consent process has been established without financial inducement.
Worked example 2 — retrospective data study: A researcher wishes to analyse de-identified hospital records of all diabetic patients over 10 years to study mortality patterns. This is low-risk observational research using existing records. The ICMR 2017 guidelines permit an IEC to grant a waiver of informed consent for secondary analysis of fully de-identified existing data, provided the research could not reasonably be done if consent were required and privacy is fully protected. The researcher still requires IEC review and approval — the waiver is IEC-granted, not researcher-declared.
Research misconduct and consequences: Fabrication of data, falsification of results, or plagiarism (FFP) are serious professional and legal violations. In India, the UGC Regulations 2018 and ICMR guidelines define research misconduct and specify sanctions ranging from retraction and ban on funding to criminal prosecution.
PIER Framework for Ethical Review of Research
SELF-CHECK
A clinical trial is being conducted at a government teaching hospital. An unexpected severe adverse event (SAE) occurs in a participant — the participant develops anaphylaxis after the first dose and requires ICU admission. What is the researcher's IMMEDIATE ethical and regulatory obligation?
A. Continue the trial and report the SAE in the final study report
B. Immediately treat the participant and report the SAE to the IEC and CDSCO within specified timeframes
C. Withdraw the participant from the study and ask the IEC for guidance within 30 days
D. Notify only the pharmaceutical company funding the trial
Reveal Answer
Answer: B. Immediately treat the participant and report the SAE to the IEC and CDSCO within specified timeframes
The first obligation is always the participant's safety — immediate treatment for anaphylaxis. The regulatory obligation under ICMR 2017 and the New Drugs and Clinical Trials Rules 2019 is to report unexpected serious adverse events expeditiously: life-threatening SAEs must be reported to the IEC and CDSCO within 24 hours (initial report) with a full report within specified further timeframes. The IEC may then review the safety signal and decide whether to suspend the trial. Delaying SAE reporting or reporting only to the sponsor is a serious regulatory violation.