Page 1 of 23
IM11.1-6 | Diabetes Foundations — SDL Guide
Learning Objectives
- Define diabetes mellitus and classify its major types with distinguishing features
- Describe the epidemiology and pathogenesis of type 1 and type 2 diabetes
- Explain the genetic and environmental determinants of diabetes
- Describe the pathogenesis and temporal evolution of microvascular and macrovascular complications
- Recognise and outline management of diabetic emergencies including DKA and HHS
INSTRUCTIONS
Diabetes mellitus is the defining metabolic epidemic of the twenty-first century and one of the highest-yield topics in final-year medicine. This module builds from first principles — definition and classification through pathogenesis, complications, and emergencies — giving you the conceptual scaffolding to handle any clinical or examination question on diabetes. Precision in diagnostic thresholds and drug mechanisms is non-negotiable at this level.
References
- Harrison's Principles of Internal Medicine, 21st ed., Ch. 403–408 — Diabetes Mellitus (textbook)
- API Textbook of Medicine, 10th ed., Ch. 51 — Diabetes Mellitus (textbook)
- Davidson's Principles and Practice of Medicine, 24th ed., Ch. 21 — Diabetes Mellitus (textbook)
- International Diabetes Federation (IDF) Atlas, 10th ed., 2021 (guideline)
- American Diabetes Association — Standards of Medical Care in Diabetes, 2024 (guideline)
Version 1.0 | NMC CBUC 2024
CLINICAL SCENARIO
Suresh, a 42-year-old software engineer from Chennai, attends his annual health check-up feeling perfectly well. He mentions mild fatigue and increased thirst over the past few months, which he attributes to work stress and summer heat. His fasting plasma glucose returns at 138 mg/dL. A repeat test the following morning gives 131 mg/dL. His HbA1c is 7.4%. He has no symptoms of hyperglycaemia — no polyuria, no weight loss — and his examination is normal. Suresh is stunned: 'But I feel fine.' Now contrast this with Kavitha, 14 years old, who is brought to the emergency department in extremis — deeply breathing, vomiting, confused, with a blood glucose of 540 mg/dL, urinary ketones 4+, and arterial pH 7.18. Her story began only three weeks ago with thirst and weight loss. Two dramatically different presentations, two different pathological mechanisms, the same diagnostic category — diabetes mellitus. Understanding why requires you to master not just the diagnostic numbers but the underlying biology of insulin deficiency, insulin resistance, and their downstream consequences.
WHY THIS MATTERS
India has the second-largest population of people with diabetes in the world — over 101 million according to the IDF 2021 Atlas — and the number is projected to rise steeply with urbanisation, dietary transition, and demographic ageing. As a final-year student and soon-to-be doctor, you will encounter diabetes in virtually every clinical setting: the general medicine ward (hyperglycaemic emergencies, infections), the outpatient clinic (chronic glycaemic management, complication screening), the surgical ward (perioperative glucose control), the obstetric unit (gestational diabetes, pre-existing diabetes in pregnancy), and the cardiology and nephrology units (diabetes-related heart disease and CKD). The NMC competency set IM11.1–11.6 requires you to operate at the KH level: not just recite facts but apply pathogenetic logic to clinical scenarios, explain complications to patients, and recognise emergencies.
RECALL
Activate your biochemistry and physiology foundation before proceeding. Insulin is a 51-amino-acid peptide hormone secreted by beta cells of the islets of Langerhans in the pancreas. It is the body's key anabolic signal: it promotes glucose uptake in muscle and fat (via GLUT-4 translocation), suppresses hepatic glucose output, promotes glycogen synthesis, inhibits lipolysis, and facilitates amino acid uptake. The counter-regulatory hormones — glucagon (alpha cells), cortisol, adrenaline, and growth hormone — oppose insulin by raising blood glucose. In the fasted state, these hormones maintain euglycaemia by driving glycogenolysis and gluconeogenesis. Glucose homeostasis depends on the precise interplay of insulin secretion and insulin sensitivity. When this system fails — from insulin deficiency, insulin resistance, or both — blood glucose rises and the clinical syndrome of diabetes emerges. Recall also the concept of glycosylated haemoglobin: glucose attaches non-enzymatically to haemoglobin A1c in proportion to the ambient glucose concentration over the preceding 8–12 weeks, making HbA1c a retrospective measure of glycaemic exposure.
Definition and Classification of Diabetes Mellitus
Diabetes mellitus is not a single disease but a group of metabolic disorders united by a common feature: chronic hyperglycaemia resulting from defects in insulin secretion, insulin action, or both. The hyperglycaemia of diabetes is not merely a laboratory abnormality — it drives a cascade of pathological changes in the vasculature, nerves, and kidneys that constitute the complications of the disease. Understanding the definition means understanding that the diagnostic thresholds are empirical boundaries derived from epidemiological studies showing the glucose level at which the risk of the specific microvascular complication of diabetic retinopathy begins to rise sharply. This is why fasting plasma glucose ≥126 mg/dL, 2-hour OGTT ≥200 mg/dL, and HbA1c ≥6.5% are not arbitrary numbers — they mark the point at which retinopathy risk inflects upward in population cohorts.
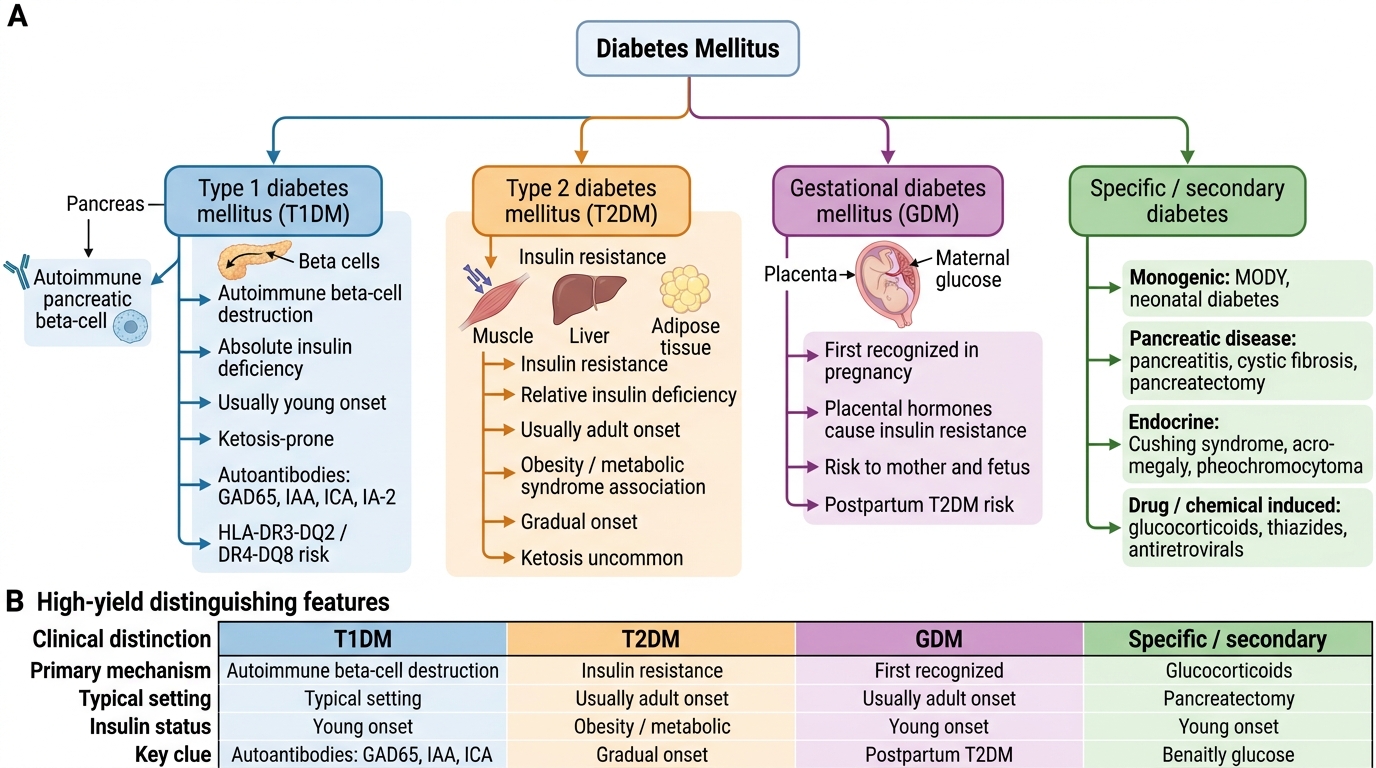
The classification of diabetes mellitus reflects aetiological heterogeneity:
- Type 1 diabetes mellitus (T1DM): Immune-mediated (autoimmune) destruction of beta cells leading to absolute insulin deficiency. Less commonly, idiopathic. Accounts for 5–10% of all diabetes. Onset typically in childhood/adolescence but can occur at any age.
- Type 2 diabetes mellitus (T2DM): Progressive defect in insulin secretion superimposed on a background of insulin resistance. Accounts for 90–95% of cases. Strong genetic and environmental (obesity, inactivity) determinants. The dominant form in India.
- Gestational diabetes mellitus (GDM): Glucose intolerance first recognised in pregnancy; resolves postpartum in most cases but confers lifetime risk of T2DM.
- Specific types (secondary diabetes): Includes maturity-onset diabetes of the young (MODY — monogenic, autosomal dominant), pancreatic diabetes (chronic pancreatitis, cystic fibrosis, haemochromatosis), endocrine causes (Cushing syndrome, acromegaly, phaeochromocytoma, glucagonoma), drug-induced (corticosteroids, thiazides, atypical antipsychotics), and mitochondrial diabetes.
In addition to established diabetes, two pre-diabetic states are recognised:
- Impaired fasting glucose (IFG): Fasting plasma glucose 100–125 mg/dL
- Impaired glucose tolerance (IGT): 2-hour OGTT glucose 140–199 mg/dL
Both IFG and IGT carry high risk of progression to T2DM and are important targets for lifestyle intervention.
⚑ AI image — pending faculty review (auto-QA score 7/10; best of 3 attempts)
Classification of Diabetes Mellitus
Pathogenesis and Epidemiology of Type 1 Diabetes
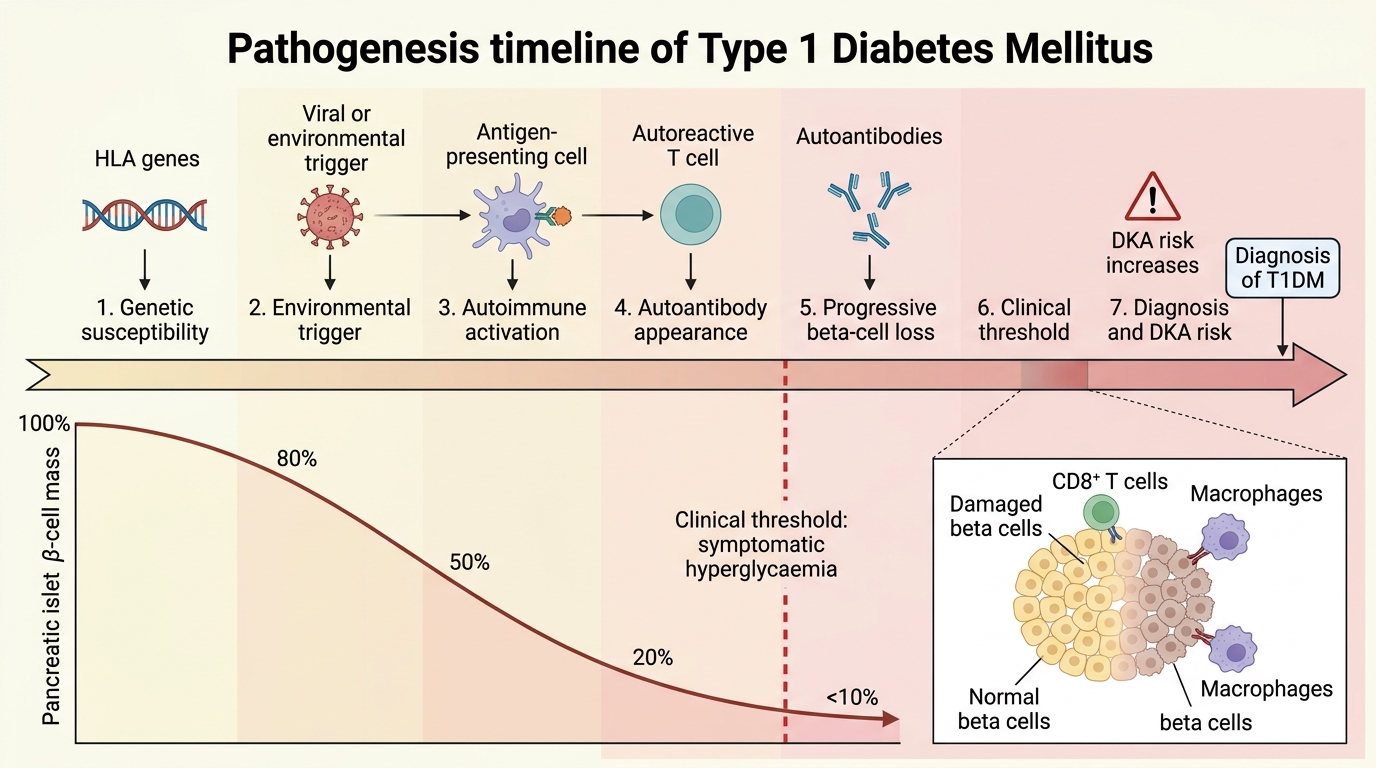
Type 1 diabetes mellitus results from autoimmune destruction of the pancreatic beta cells, leading to absolute deficiency of insulin. The process is initiated in genetically susceptible individuals by an environmental trigger — most commonly an enteric viral infection (especially Coxsackievirus B4) or, in hypothesis, early dietary antigens — that activates autoreactive CD4+ and CD8+ T lymphocytes targeting islet-specific proteins. The primary autoantigens include glutamic acid decarboxylase (GAD65), insulin autoantibodies (IAA), islet cell antibodies (ICA), and IA-2 (islet antigen 2, a protein tyrosine phosphatase). These autoantibodies are detectable in the serum months to years before clinical symptoms appear, during a prolonged pre-clinical phase of progressive beta-cell destruction. Clinical diabetes manifests only when approximately 80–90% of beta-cell mass has been destroyed, at which point residual insulin secretion is insufficient to prevent hyperglycaemia even in the basal state.
Genetic determinants of T1DM are dominated by the HLA (human leucocyte antigen) region on chromosome 6p21, which accounts for 40–50% of the genetic susceptibility. The highest-risk HLA haplotypes are DR3-DQ2 and DR4-DQ8; individuals carrying both haplotypes (HLA-DR3/DR4 heterozygotes) have a risk of approximately 1 in 20. Concordance in identical twins is only 30–50%, demonstrating that environmental factors are essential co-determinants. A dozen or more non-HLA susceptibility loci have been identified, including the insulin gene region (INS VNTR) on chromosome 11 and CTLA-4 (a T-cell negative regulatory molecule), collectively contributing the remaining genetic risk.
Epidemiology: T1DM has an annual incidence of approximately 15–30 per 100,000 children under 15 years in high-income countries; India has a lower incidence (around 3–5 per 100,000) but a large absolute number of cases due to population size. The peak age at diagnosis is 5–7 years (often coinciding with school entry and new viral exposure) and again at 10–14 years (pubertal period). The incidence of T1DM has been rising in many countries, particularly in children under 5, at a rate unexplained by genetic drift — implicating accelerating environmental change (the 'overload hypothesis' and 'hygiene hypothesis').
Clinical evolution: The pre-clinical phase can last years. Clinical onset is typically abrupt — days to weeks of polyuria, polydipsia, polyphagia, and weight loss, often precipitated by the stress of an intercurrent illness. Without insulin replacement, ketogenesis is unchecked and diabetic ketoacidosis (DKA) ensues. After diagnosis and insulin initiation, a honeymoon period (partial remission) of weeks to months is common in some patients, as residual beta-cell function is temporarily preserved by reducing the glucotoxic stress on surviving cells. This phase ends as immune destruction progresses to completion.
Pathogenesis Timeline of Type 1 Diabetes Mellitus
Pathogenesis and Epidemiology of Type 2 Diabetes
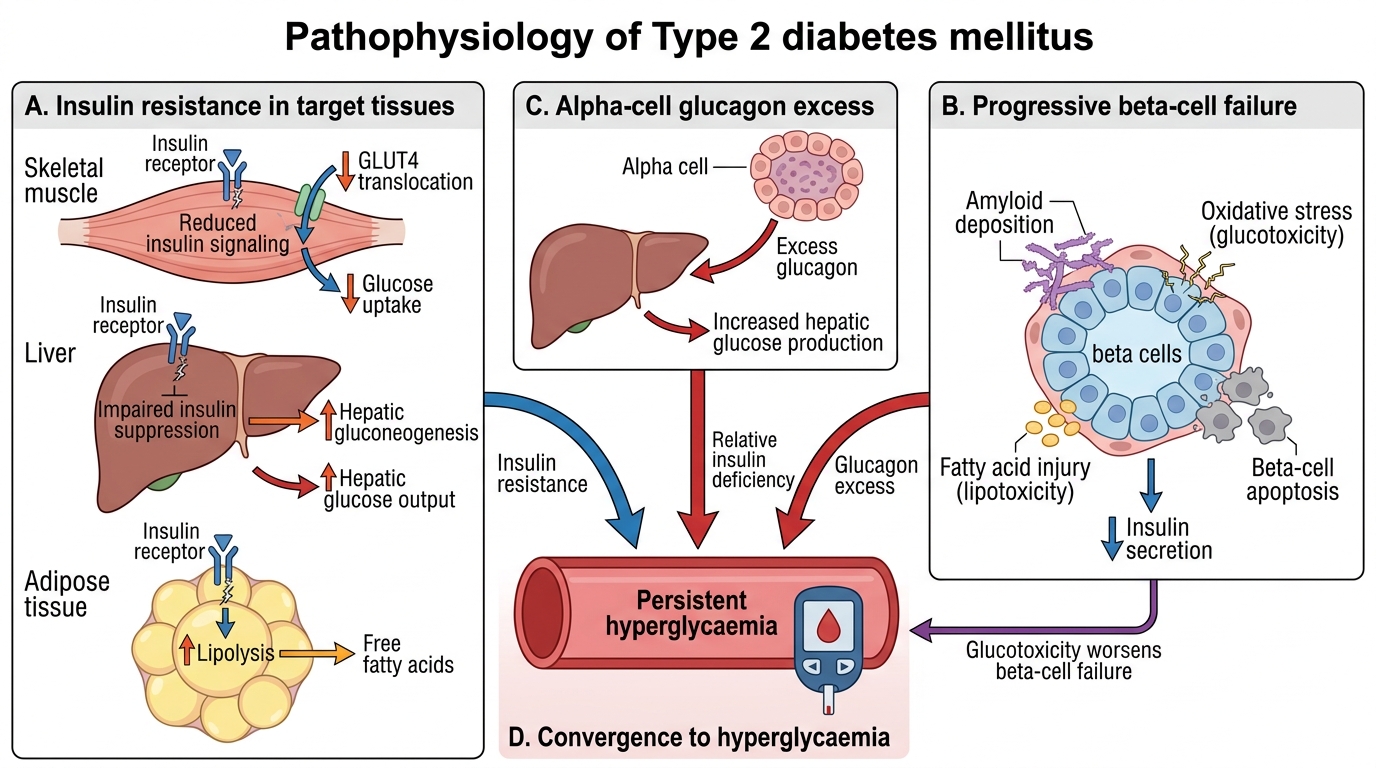
Type 2 diabetes mellitus is a progressive metabolic disorder defined by two core defects operating in parallel: peripheral insulin resistance (particularly in skeletal muscle, adipose tissue, and liver) and progressive beta-cell secretory failure. Neither defect alone is sufficient to cause diabetes — both must be present. This insight, articulated by the 'twin defect' model, explains why some obese insulin-resistant individuals never develop T2DM (their beta cells compensate adequately) while others progress inexorably despite modest insulin resistance (their beta-cell reserve is limited from the outset by genetic factors).
Insulin resistance arises from a complex interaction of genetic predisposition, excess adiposity (especially visceral/ectopic fat), physical inactivity, and ageing. Visceral adipocytes release excess free fatty acids (FFA) into the portal circulation, which impair hepatic insulin signalling and drive excess hepatic glucose output. In skeletal muscle, intramyocellular lipid accumulation and mitochondrial dysfunction impair insulin-stimulated GLUT-4 translocation. Adipokine dysregulation — excess TNF-α, IL-6, and resistin, with deficient adiponectin — amplifies the inflammatory component of insulin resistance. The ectopic fat hypothesis proposes that fat deposition in the liver and pancreas (visible on MRI as pancreatic fat) directly impairs both hepatic insulin sensitivity and beta-cell function, providing the mechanistic link between visceral obesity and T2DM.
Beta-cell failure in T2DM is both quantitative (progressive loss of beta-cell mass by apoptosis, estimated at ~40–60% reduction at diagnosis) and qualitative (impaired glucose-stimulated insulin secretion, altered insulin pulse pattern, impaired first-phase insulin release). Amyloid deposition in the islets — from islet amyloid polypeptide (IAPP, also called amylin, co-secreted with insulin) — is a histological hallmark of T2DM pancreas and may contribute to beta-cell toxicity. Glucotoxicity (chronic hyperglycaemia impairing insulin secretion) and lipotoxicity (excess FFA impairing beta-cell function) create a vicious cycle accelerating beta-cell decline.
Genetic determinants of T2DM: Unlike T1DM where single HLA loci dominate, T2DM is highly polygenic, with >250 susceptibility loci identified by GWAS. Concordance in identical twins is 70–90%, reflecting a stronger genetic contribution than T1DM, yet no single gene is necessary or sufficient. Indians and South Asians have a particular vulnerability to T2DM at lower BMI thresholds than Caucasians — a phenomenon attributed to the 'thin-fat Indian' phenotype described by Yajnik: Indian neonates and adults have higher body fat percentage and visceral adiposity relative to their weight compared to European counterparts, conferring greater metabolic risk at lower BMI. This has prompted India-specific BMI thresholds for obesity screening (≥23 kg/m² for overweight, ≥25 kg/m² for obesity).
Epidemiology: India has approximately 101 million people with diabetes (IDF 2021), with T2DM accounting for over 95% of cases. Prevalence is highest in urban areas and southern states. The economic impact is enormous — estimated annual direct medical cost of diabetes in India exceeds USD 2 billion — with a disproportionate burden on the working-age population, directly impacting productivity, healthcare expenditure, and national GDP. The IDF projects India's diabetic population will reach 135 million by 2045.
Clinical evolution of T2DM: Unlike T1DM, the onset of T2DM is insidious. Many patients are asymptomatic for years; in India, approximately 57% of diabetes cases remain undiagnosed. The clinical course follows a natural history: insulin resistance → compensatory hyperinsulinaemia → impaired fasting glucose (IFG) → impaired glucose tolerance (IGT) → overt T2DM → progressive complications. Even before formal diagnosis, microvascular complications (particularly retinopathy) may already be present, underscoring the importance of opportunistic screening in high-risk populations.
Twin Defects in Type 2 Diabetes Mellitus
SELF-CHECK
An 18-year-old presents with 3 weeks of polyuria, polydipsia, and 4 kg weight loss. His fasting blood glucose is 320 mg/dL and urinary ketones are 3+. Which of the following best characterises the primary pathological mechanism in this patient?
A. Peripheral insulin resistance with compensatory hyperinsulinaemia
B. Amyloid deposition in the islets causing progressive beta-cell apoptosis
C. Autoimmune destruction of beta cells leading to absolute insulin deficiency
D. Excess glucagon secretion from alpha cells secondary to impaired hepatic insulin signalling
Reveal Answer
Answer: C. Autoimmune destruction of beta cells leading to absolute insulin deficiency
The presentation — young patient, acute onset, significant weight loss, ketonuria — is characteristic of type 1 diabetes mellitus. The primary mechanism is autoimmune (CD4+/CD8+ T-cell mediated) destruction of pancreatic beta cells leading to absolute insulin deficiency. Without insulin, lipolysis is unchecked and ketone bodies accumulate, explaining the ketonuria. Peripheral insulin resistance and amyloid deposition are mechanisms in T2DM. Alpha-cell excess glucagon is a contributing factor in both T1DM and T2DM but is not the primary aetiology.