Page 3 of 13
IM20.1-2 | Seizure Disorder Foundations and Diagnosis — SDL Guide (Part 3)
Epilepsy Syndromes — Clinical Recognition
An epilepsy syndrome is a specific electroclinical constellation — a characteristic combination of seizure type(s), EEG features, age of onset, neuroimaging findings, and sometimes genetic background — that recurs predictably and has implications for prognosis and treatment. Recognising a syndrome allows more precise prognostic counselling and optimises AED selection. The following syndromes are high-yield for the general physician.
Provided image
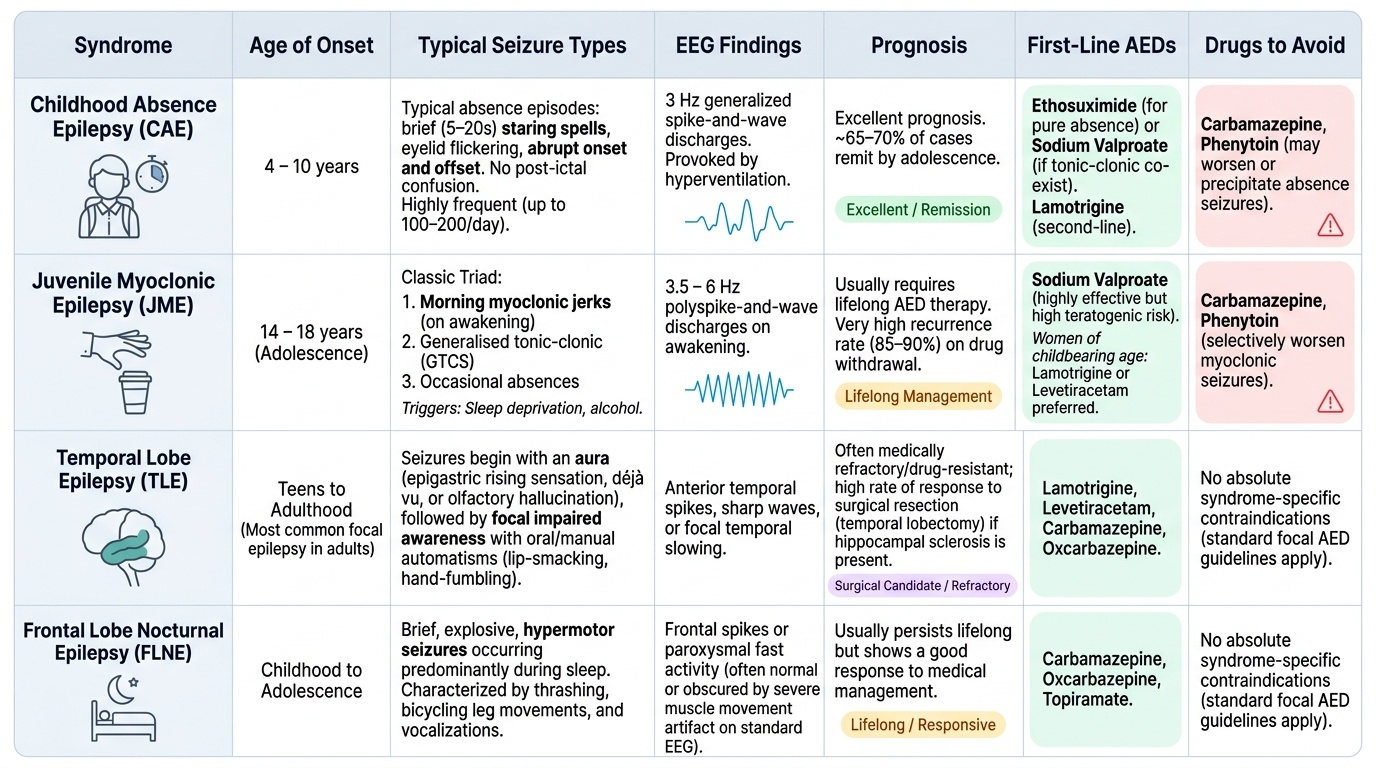
Childhood Absence Epilepsy (CAE): Onset between ages 4 and 10 years. Seizures are very frequent (up to 100–200 per day) typical absence episodes — brief (5–20 seconds) staring with eyelid flicker, abrupt onset and offset, and no post-ictal confusion. The pathognomonic EEG finding is 3 Hz generalised spike-and-wave discharges provoked by hyperventilation. The seizures have an excellent prognosis — approximately 65–70% remit by adolescence. First-line treatment: ethosuximide (for pure absence without tonic-clonic) or sodium valproate (when tonic-clonic co-exist); lamotrigine is second-line. Carbamazepine and phenytoin are contraindicated — they may worsen or precipitate absence seizures.
Juvenile Myoclonic Epilepsy (JME): Onset in adolescence (14–18 years). The classic triad is: (1) morning myoclonic jerks on awakening (often causing the patient to drop their cup of coffee or toothbrush — a specific history worth asking), (2) generalised tonic-clonic seizures, and (3) occasional absence seizures. Sleep deprivation and alcohol are powerful triggers. EEG shows polyspike-and-wave at 3.5–6 Hz on awakening. JME usually requires lifelong AED therapy — recurrence on drug withdrawal is very high (85–90%). Sodium valproate is the most effective drug but carries high teratogenicity risk in women. In women of childbearing age, lamotrigine or levetiracetam are preferred (see management module). Carbamazepine and phenytoin are contraindicated in JME — they selectively worsen myoclonic seizures while having no effect on the syndrome.
Temporal Lobe Epilepsy (TLE): The most common form of focal epilepsy in adults. The most frequent substrate is hippocampal sclerosis — mesial temporal sclerosis (MTS). Seizures begin with an aura (most commonly an epigastric rising sensation, déjà vu, or olfactory hallucination), followed by a focal impaired awareness seizure with oral and manual automatisms (lip-smacking, chewing, hand-picking), head version, and post-ictal confusion lasting several minutes. Secondary generalisation can occur. TLE is the most surgically remediable epilepsy — anterior temporal lobectomy achieves seizure freedom in 60–70% of carefully selected patients with MTS.
Febrile Seizures: Occur in 2–5% of children aged 6 months to 6 years, by definition in the context of fever (temperature >38°C) without CNS infection or metabolic cause. Simple febrile seizures (generalisede, <15 minutes, single in a 24-hour period) are benign and do not require AED therapy. Complex febrile seizures (focal, >15 minutes, multiple within 24 hours, or with post-ictal deficit) carry a higher risk of subsequent epilepsy and warrant investigation.
Neonatal Seizures: Common in the first month of life; most are symptomatic (hypoxic-ischaemic encephalopathy, hypoglycaemia, hypocalcaemia, pyridoxine deficiency, intracranial haemorrhage, metabolic disease). Subtle forms (ocular deviation, lip-smacking, swimming movements) are easily missed. Phenobarbital is first-line.
SELF-CHECK
A 16-year-old girl is referred because of episodes of bilateral arm jerking in the morning, usually within 30 minutes of waking. She also describes two episodes of losing consciousness and falling, both occurring within an hour of waking on mornings after parties. She was given a prescription for carbamazepine by another doctor. What is the MOST APPROPRIATE management change?
A. Increase the dose of carbamazepine
B. Add levetiracetam to carbamazepine
C. Stop carbamazepine and start sodium valproate or levetiracetam
D. Refer for EEG-video telemetry before changing any medication
Reveal Answer
Answer: C. Stop carbamazepine and start sodium valproate or levetiracetam
This is classic Juvenile Myoclonic Epilepsy (JME) — morning myoclonic jerks, sleep-deprivation and post-party GTCS, adolescent onset. Carbamazepine is CONTRAINDICATED in JME because it selectively worsens myoclonic seizures while providing no benefit for the syndrome's generalised seizure types. It must be stopped. Sodium valproate is the most effective first-line treatment; levetiracetam is the preferred alternative in women of childbearing age (given valproate's teratogenicity and PCOS risk). Increasing carbamazepine would worsen the myoclonus. Adding levetiracetam without removing carbamazepine still leaves the harmful drug in place. An EEG is appropriate but is not the priority — the dangerous drug must be stopped first, particularly since the clinical diagnosis of JME is already clear.
Self-Assessment: Applying the Diagnostic Framework
At this point in the module you have covered the precise definitions of seizure, convulsion, and epilepsy; the ILAE 2017 classification by onset type; the six aetiological categories with Indian context (neurocysticercosis, cerebral malaria); the pathophysiology of seizure generation (PDS, excitation-inhibition imbalance, T-type Ca²⁺ channels in absence, GABA-A internalisation in status epilepticus); clinical evaluation including differentials (syncope, PNES, TIA, hypoglycaemia); and the diagnostic investigations (blood tests, EEG pattern recognition, MRI protocol, CSF). The self-assessment scenarios below ask you to apply this framework to clinical vignettes at the NMC IM20.1–IM20.2 competency level.
Scenario A: A 22-year-old male labourer from rural Bihar presents to a district hospital with two witnessed tonic-clonic seizures in the past month. CT brain shows a small hyperdense calcified nodule in the right parietal lobe with surrounding oedema. There is no fever. Serum electrolytes and glucose are normal. He has no family history of epilepsy.
Analysis: Single ring-enhancing or calcifying lesion in a young adult from an area where pork is consumed = neurocysticercosis (NCC) until proven otherwise. This is a structural aetiology. The seizures are unprovoked (the cysticercal granuloma is a permanent structural lesion, not a transient provocation). Two seizures fulfil the ILAE definition of epilepsy. Workup: CT + cysticercal serology (IgG ELISA), stool examination for Taenia ova. Anti-parasitic therapy (albendazole ± praziquantel) combined with corticosteroids (to suppress the inflammatory oedema) alongside AED treatment. Do NOT label this as idiopathic epilepsy.
Scenario B: A 34-year-old woman with known lupus erythematosus develops acute-onset headache, psychosis, and three focal seizures over 36 hours. MRI brain shows FLAIR hyperintensities in bilateral temporal and frontal regions without mass effect. Blood glucose and electrolytes are normal.
Analysis: Subacute multi-area encephalopathy with psychosis + seizures in a lupus patient suggests either CNS lupus or autoimmune encephalitis (anti-NMDAR or anti-LGI1 most likely). Aetiology is immune. Investigations: CSF for cells, protein, oligoclonal bands; serum and CSF anti-neuronal antibodies (anti-NMDAR, anti-LGI1, anti-CASPR2); lupus serology (dsDNA, complement). Management: immunosuppression (methylprednisolone, IVIG), treat the underlying lupus, AEDs for seizure control.
Scenario C: A 6-year-old girl's teacher reports that she stares blankly for 5–10 seconds, 10–15 times a day, during class. Her parents thought she was daydreaming. Her development is normal. Her paediatric neurologist finds that hyperventilation for 30 seconds during the consultation produces one of these episodes, with a blank stare and eyelid flicker.
Analysis: Brief, frequent, non-convulsive episodes with abrupt onset and offset, provoked by hyperventilation, in a 6-year-old with normal development = childhood absence epilepsy. The expected EEG finding is 3 Hz spike-and-wave. First-line treatment is ethosuximide (if absence-only) or valproate (if GTCS co-exist or risk is considered). Carbamazepine is absolutely contraindicated and must never be prescribed.
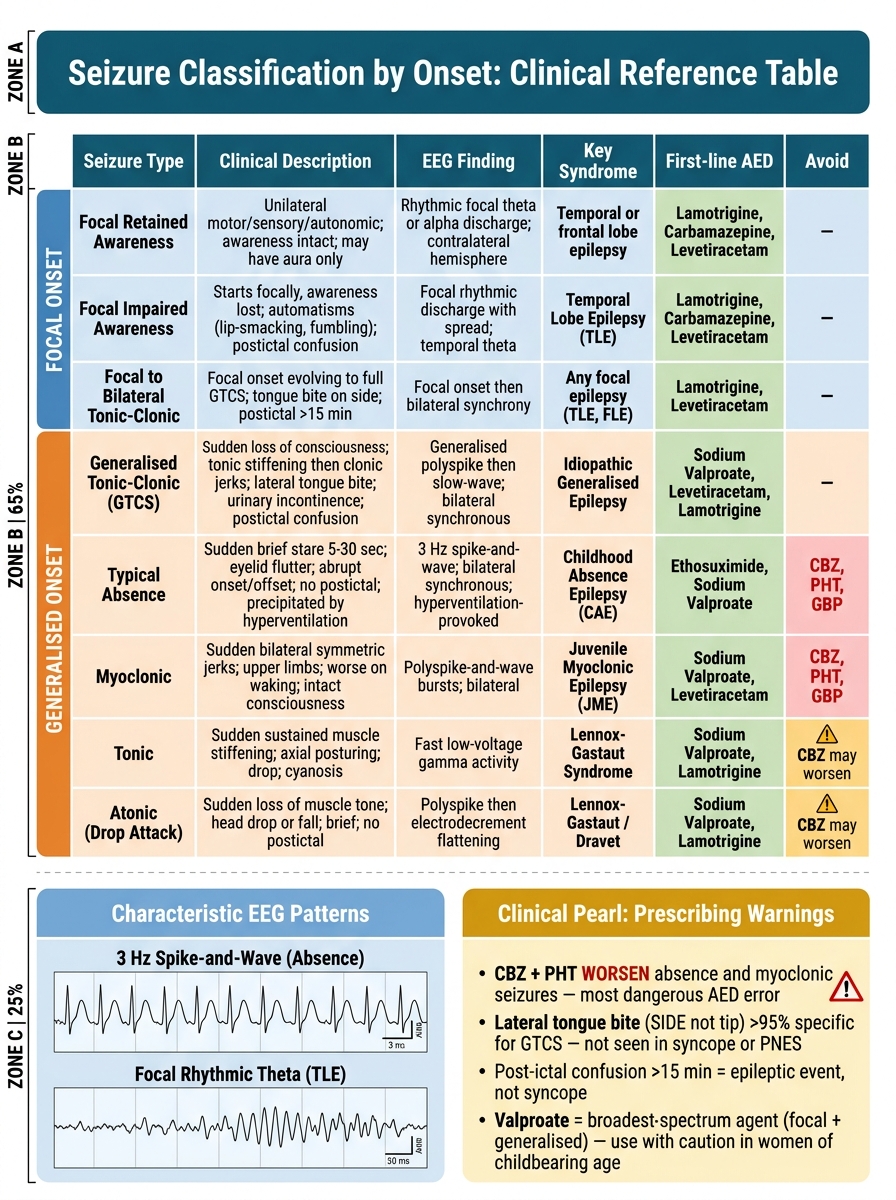
Seizure Classification by Onset — Clinical Reference Table with AED Selection Guide
CLINICAL PEARL
Two clinical pearls are essential for the diagnostic evaluation of seizures.
First: lateral tongue biting is highly specific for a generalised tonic-clonic epileptic seizure (specificity >95%) and virtually never occurs in vasovagal syncope or PNES. The bite is on the side of the tongue, not the tip. Cheek or lip biting is less specific. Urinary incontinence is also more common with epileptic GTCS than syncope, but it can occur in syncope. Post-ictal confusion lasting >5–15 minutes strongly favours an epileptic event.
Second: carbamazepine and phenytoin worsen absence and myoclonic seizures — this is one of the most dangerous and common prescribing errors in epilepsy. A patient with JME or childhood absence epilepsy prescribed carbamazepine will typically get worse: myoclonic jerks intensify and absence seizures become more frequent. Always classify the seizure type before choosing an AED. If the seizure type is unclear, sodium valproate is the broadest-spectrum first-line agent (effective across focal and generalised types) but requires careful consideration in women of childbearing age.