Page 2 of 13
IM20.1-2 | Seizure Disorder Foundations and Diagnosis — SDL Guide (Part 2)
Pathophysiology of Seizure Generation
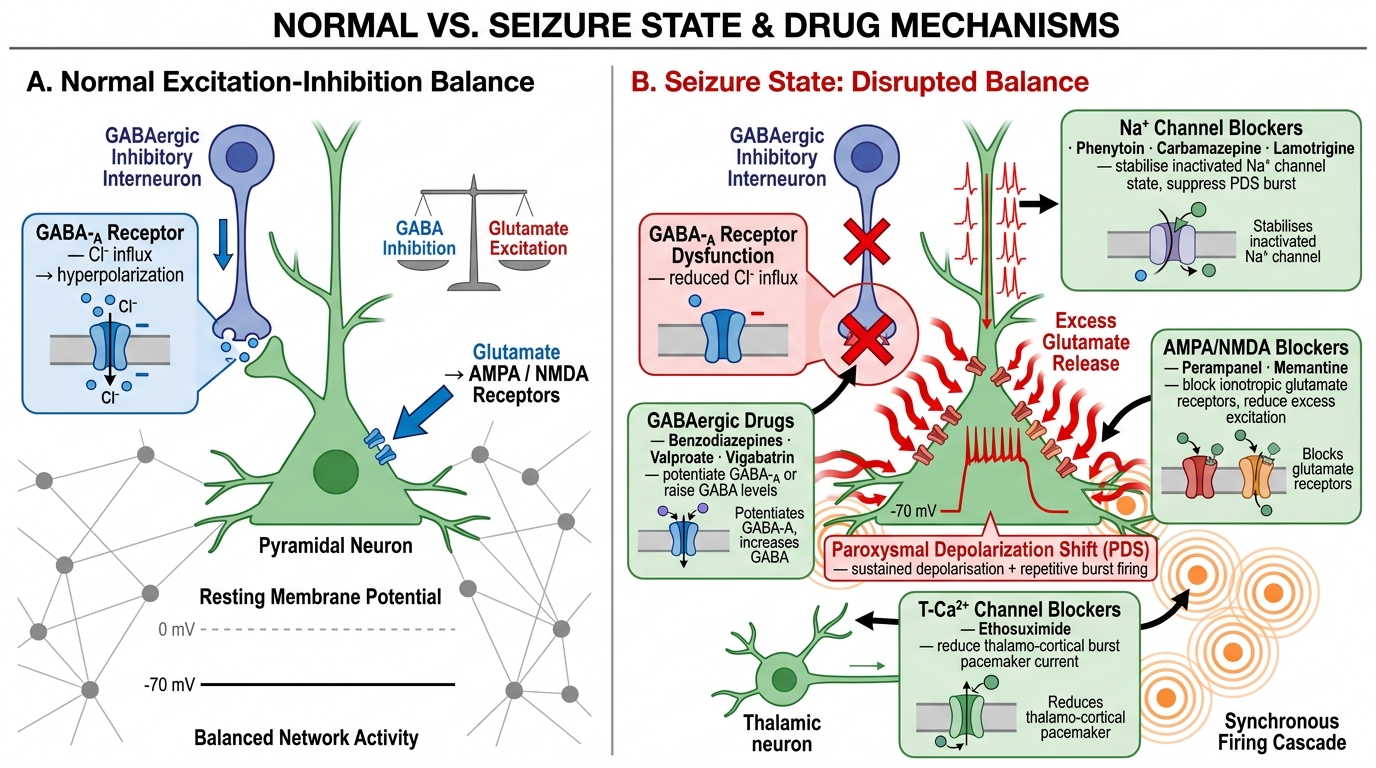
Understanding the mechanism by which a seizure is generated illuminates why certain metabolic and pharmacological conditions are epileptogenic, and why particular AEDs are effective against specific seizure types. The fundamental abnormality in all seizures is an imbalance between excitation and inhibition in a cortical or subcortical neuronal network, resulting in paroxysmal depolarisation shift (PDS) — the electrophysiological hallmark of the epileptic neuron.
The paroxysmal depolarisation shift consists of a large, sustained membrane depolarisation (lasting 50–200 ms) during which the neuron fires repetitively at very high frequency, followed by a prolonged afterhyperpolarisation. At the single-neuron level, the PDS is mediated by Ca²⁺ influx through L-type (high-voltage-activated) calcium channels and persistent Na⁺ current through voltage-gated sodium channels. When the PDS spreads to a sufficient population of neurons — via synaptic transmission and gap junctions — a focal ictal discharge occurs. Whether this focal discharge remains local (focal seizure) or propagates to engage bilateral networks (generalised or secondarily generalised seizure) depends on the balance of inhibitory (GABAergic) and excitatory (glutamatergic) circuits surrounding the focus, the integrity of commissural fibres, and the excitability state of the reticular formation.
Three principal mechanisms disrupt the excitation-inhibition balance:
- Reduced GABAergic inhibition: GABA-A receptor channels conduct chloride ions, hyperpolarising the neuron. Reduced GABA synthesis (pyridoxine deficiency), GABA receptor downregulation (prolonged benzodiazepine use followed by abrupt withdrawal), GABA receptor blockade (penicillin at high doses, isoniazid — which inhibits pyridoxal phosphate, the GABA cofactor), or inherited GABA subunit mutations all lower the seizure threshold.
- Excessive glutamatergic excitation: Glutamate acting through AMPA and NMDA receptors drives the PDS. Hypoxia, hypoglycaemia, and hyperthermia all impair glutamate reuptake by astrocytes and drive receptor overactivation. NMDA receptors additionally require Mg²⁺ channel blockade for activation; hypomagnesaemia removes this block and promotes seizure generation — the mechanism of eclamptic seizures.
- Intrinsic neuronal membrane channel dysfunction: Ion channel mutations alter the threshold for action potential generation. Loss-of-function mutations in voltage-gated sodium channel subunits (SCN1A in Dravet syndrome — loss of function in inhibitory interneurons paradoxically increases network excitability), and gain-of-function mutations in sodium or calcium channels directly lower the seizure threshold.
The absence seizure is generated by a distinct mechanism: abnormal thalamo-cortical oscillation at 3 Hz, driven by T-type (low-voltage-activated) calcium channels in thalamic relay and reticular neurons. This explains why ethosuximide — which selectively blocks T-type Ca²⁺ channels — is specifically effective for typical absence seizures but ineffective for focal or tonic-clonic seizures.
Status epilepticus represents a failure of the normal seizure-termination mechanisms. As a seizure continues beyond 5 minutes, GABA-A receptors are internalised (removed from the synapse), progressively reducing GABAergic inhibition and making benzodiazepines less effective over time — the pharmacological basis for the time-sensitive stepwise management discussed in the companion module.
Cortical Excitation-Inhibition Balance and Seizure Pathophysiology with Drug Targets
SELF-CHECK
A 30-year-old man has a witnessed tonic-clonic convulsion. He reports that just before the convulsion, his right hand started shaking for about 15 seconds, then 'the shaking spread up his arm and he lost awareness'. On examination post-ictally, he has temporary weakness of the right arm. Which of the following BEST explains these clinical features?
A. Generalised onset tonic-clonic seizure due to genetic epilepsy
B. Typical absence seizure with secondary spread
C. Focal onset seizure with secondary bilateral spread (focal to bilateral tonic-clonic seizure) with Todd's palsy
D. Psychogenic non-epileptic seizure (PNES)
Reveal Answer
Answer: C. Focal onset seizure with secondary bilateral spread (focal to bilateral tonic-clonic seizure) with Todd's palsy
The sequential onset (right hand jerking → arm → loss of awareness → bilateral tonic-clonic) is the classic 'Jacksonian march' pattern of a focal motor seizure with secondary generalisation — now termed 'focal to bilateral tonic-clonic seizure' in ILAE 2017 terminology. The post-ictal right arm weakness is Todd's palsy — a transient focal weakness lasting minutes to hours after a focal motor seizure, reflecting post-ictal cortical depression in the motor strip. This indicates a focal structural cause (contra-lateral to the weak limb) requiring MRI. Generalised onset epilepsy (genetic) does not have a focal onset aura or post-ictal focal deficit. Absence seizures do not spread to bilateral tonic-clonic events. PNES would not produce a stereotyped Jacksonian march.
Clinical Evaluation of the First Seizure
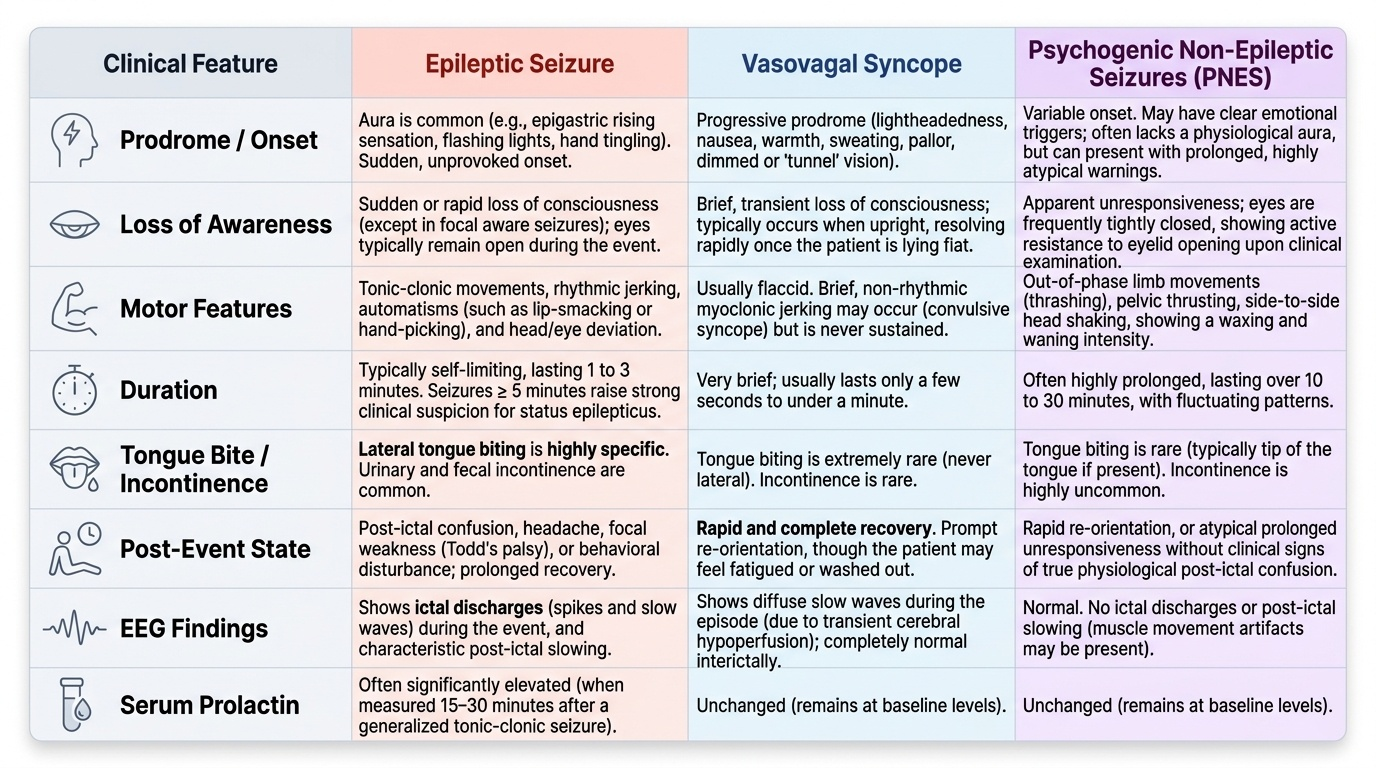
The clinical evaluation after a first seizure has two parallel goals: first, confirm that what occurred was indeed a seizure (and not a mimic), and second, identify any underlying aetiology — particularly a life-threatening or reversible cause. The history from witnesses is the most powerful diagnostic tool available, since the patient is typically post-ictal and amnestic for the event itself. No investigation can substitute for a detailed, structured seizure history.
Provided image
The history should establish: (1) the onset — was there a warning (aura)? An aura is in fact a focal seizure preceding generalisation; its character localises the seizure onset zone (epigastric rising sensation → mesial temporal; flashing lights → occipital cortex; tingling of the hand → contralateral sensorimotor cortex). (2) The ictal phase — was there loss of consciousness, motor activity (which limbs first, tonic vs clonic, rhythmic vs non-rhythmic), automatisms (lip-smacking, chewing, hand-picking in temporal lobe seizures), head and eye deviation, vocalisation, incontinence, tongue biting? Lateral tongue biting is highly specific for tonic-clonic seizures. (3) Duration — seizures lasting >5 minutes raise the suspicion of developing status epilepticus. (4) Post-ictal phase — duration and character of recovery (confusion, headache, focal weakness suggesting Todd's palsy, behavioural disturbance in temporal lobe seizures). (5) Precipitants — sleep deprivation (common trigger for JME), alcohol (withdrawal is a powerful trigger), photic stimulation, fever, medications (tramadol, theophylline, fluoroquinolones, bupropion, imipenem all lower the seizure threshold). (6) Previous episodes — the two cases in the hook illustrate why every first apparent tonic-clonic seizure requires a careful history for prior 'minor' episodes that may represent preceding focal seizures.
The differential diagnosis of seizures — events that mimic seizures — must be actively considered. The most important mimics in clinical practice are: syncope (vasovagal, cardiac, or orthostatic — brief myoclonic jerks during syncope can be misinterpreted as tonic-clonic seizure; syncope has a prodrome of lightheadedness, nausea, pallor; rapid and complete recovery; no tongue bite or incontinence; EEG during syncopal episode shows diffuse slow waves, not ictal discharge); PNES (psychogenic non-epileptic seizures — also called dissociative seizures; characterised by waxing-and-waning course, pelvic thrusting, eye closure, absence of cyanosis despite prolonged 'convulsion', resistance to eye opening, intact corneal reflex during event; ictal prolactin NOT reliably elevated unlike in epileptic seizures); transient ischaemic attack (TIA) (sudden focal neurological deficit rather than ictal motor activity; typically negative symptoms — weakness, sensory loss — rather than positive symptoms — jerking); migraine with aura (similar aura phenomenology to occipital epilepsy but migraine aura spreads slowly over 5–20 minutes while ictal spread is seconds); hypoglycaemia (can produce focal neurological deficit or convulsions, immediately corrected by glucose); movement disorders and REM sleep behaviour disorder (abnormal nocturnal motor events).
Diagnostic Tests in Epilepsy
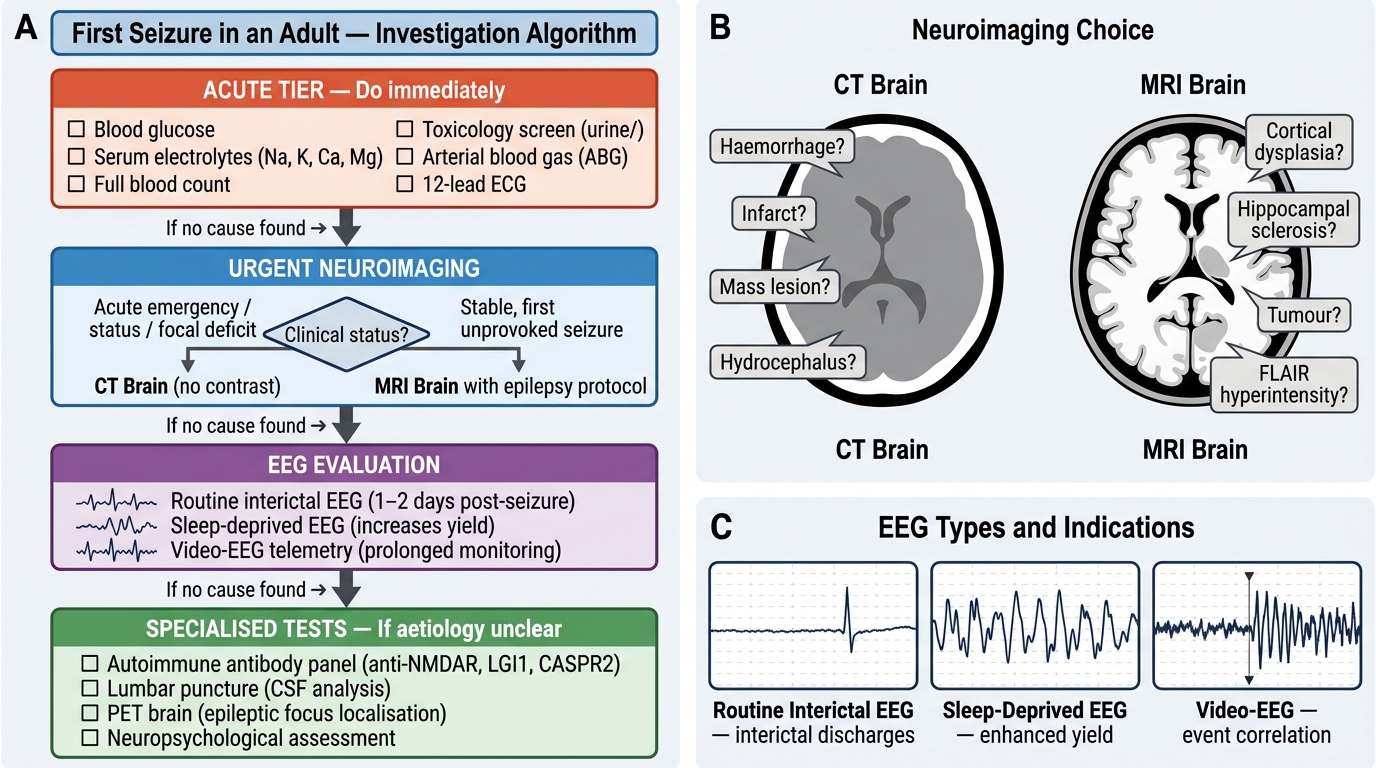
The investigation of a first seizure is directed by the clinical probability of epilepsy, the urgency of identifying a structural or metabolic cause, and the need for accurate classification to guide treatment. No single test diagnoses epilepsy — the diagnosis remains fundamentally clinical, supported and refined by a layered set of investigations. A useful mental model is to think of investigations in three tiers: first, the acute metabolic screen (immediate, bedside or rapid-turnaround tests that identify provoked seizures requiring treatment of the underlying cause rather than an AED); second, the electrophysiological investigation (EEG, which characterises the seizure type, identifies a syndrome, and distinguishes epileptic from non-epileptic events); and third, structural neuroimaging (MRI or CT, which identifies or excludes an underlying anatomical substrate). Each tier answers a specific question: 'Is this a metabolic crisis?', 'What type of epilepsy is this?', and 'Is there a lesion driving this?'. Investigations are ordered in sequence or in parallel depending on clinical urgency — the acutely seizing patient needs the metabolic screen simultaneously with imaging; the stable outpatient presenting with a first witnessed convulsion can be worked up in a stepwise manner.
Urgent blood tests (always in the acute setting):
- Blood glucose — hypoglycaemia (<2.8 mmol/L or <50 mg/dL) is an immediate, reversible cause; it must be checked and corrected before any other workup.
- Serum electrolytes: sodium (hyponatraemia <125 mEq/L is epileptogenic), calcium (hypocalcaemia causes tetany and seizures), magnesium, urea, and creatinine (uraemic seizures in CKD). Correcting these metabolic derangements is the primary treatment — AEDs are not indicated for purely provoked metabolic seizures.
- Full blood count, liver function tests: identify systemic disease, inform AED choice (hepatic metabolism of most AEDs).
- Arterial blood gas: lactic acidosis (prolonged seizure), respiratory acidosis (peri-ictal hypoventilation).
- Serum prolactin: rises transiently (typically within 10–20 minutes of seizure onset and normalises by 60 minutes) after generalised tonic-clonic and complex focal seizures but NOT after absence seizures or PNES. An elevated post-ictal prolactin (>2× upper limit of normal) supports an epileptic event; a normal prolactin does not exclude one.
- Toxicology screen: drug overdose (stimulants, tramadol, tricyclics, bupropion) and alcohol withdrawal.
- In appropriate contexts: AED levels (in known epilepsy with breakthrough seizures — confirms non-compliance or toxicity), anti-NMDAR/anti-LGI1 antibodies (clinical suspicion of autoimmune encephalitis), blood cultures and CSF (if meningitis/encephalitis suspected).
Electroencephalography (EEG):
The EEG is the most important specific investigation in epilepsy. It records the electrical activity of the superficial cortex via scalp electrodes, typically over 20–40 minutes. A routine interictal EEG (recorded between seizures) may show epileptiform discharges — spikes, sharp waves, or spike-and-wave complexes — which increase the probability of epilepsy and help classify the syndrome. However, a normal EEG does not exclude epilepsy: approximately 50% of patients with epilepsy have a normal interictal EEG on a single recording. Yield is increased by sleep deprivation (captures drowsy and early sleep EEG), activation procedures (hyperventilation provokes absence seizures; photic stimulation provokes photosensitive epilepsy), and repeated recordings. Specific patterns:
- 3 Hz generalised spike-and-wave: typical absence epilepsy (childhood absence, juvenile absence).
- Focal interictal spike/sharp wave over the temporal region: temporal lobe epilepsy.
- Polyspike-and-wave (3.5–6 Hz) on awakening: juvenile myoclonic epilepsy.
- Hypsarrhythmia (chaotic high-amplitude slow waves with multifocal spikes): infantile spasms (West syndrome).
- Slow spike-and-wave (<3 Hz) with background abnormality: Lennox-Gastaut syndrome.
Video-EEG is the gold standard for characterising seizure semiology and correlating with EEG changes; it is essential for pre-surgical evaluation and for definitively distinguishing epileptic seizures from PNES.
Neuroimaging:
MRI brain (with epilepsy protocol) is the preferred imaging modality for structural cause identification. It should be performed in all adults with new-onset epilepsy and in any patient with focal onset seizures, neurological signs, seizures starting after age 25 (when a structural lesion is more likely than genetic epilepsy), or inadequate response to AEDs. The epilepsy MRI protocol uses specific sequences: coronal T2/FLAIR oblique to the hippocampus (identifies hippocampal sclerosis — the most common surgically remediable epilepsy cause), gadolinium-enhanced T1 (tumour, inflammation), T2* or susceptibility-weighted imaging (microbleeds, cavernoma, old haemorrhage), and FLAIR (cortical dysplasia, encephalitis, post-ictal signal). CT brain is useful in the acute setting (subarachnoid haemorrhage, haematoma, acute meningitis, gross tumour) but has poor sensitivity for most epileptogenic substrates compared to MRI. CT is appropriate when MRI is contraindicated or unavailable.
Lumbar puncture (CSF analysis):
Mandatory when meningitis, encephalitis, or subarachnoid haemorrhage is suspected and CT has excluded a space-occupying lesion with raised ICP. Should be performed when there is fever, meningism, persistent unexplained confusion, or first seizure in an immunocompromised patient. CSF findings in common causes: bacterial meningitis (high polymorphs, high protein, very low glucose); viral encephalitis (lymphocytes, mildly elevated protein, normal or mildly low glucose, viral PCR); TB meningitis (lymphocytes, high protein, very low glucose, AFB smear, TB-PCR, ADA). Anti-NMDAR antibody can also be measured in CSF (higher sensitivity than serum in some studies).
Neuropsychological assessment is part of the pre-surgical evaluation in refractory epilepsy and provides a functional localisation of language and memory relevant to temporal lobe surgery.
Investigation Algorithm for First Seizure in an Adult
SELF-CHECK
A 19-year-old woman is brought to casualty after a first episode of generalised tonic-clonic seizure. She is post-ictal. Blood glucose is 88 mg/dL. Which investigation is MOST important to perform NEXT, before ordering an EEG or MRI?
A. Serum prolactin (to confirm the seizure was epileptic)
B. Serum electrolytes including sodium, calcium, and magnesium
C. Urine toxicology screen
D. CT brain without contrast
Reveal Answer
Answer: B. Serum electrolytes including sodium, calcium, and magnesium
After glucose (already checked and normal), serum electrolytes — specifically sodium, calcium, and magnesium — are the next most important urgent tests. Hyponatraemia, hypocalcaemia, and hypomagnesaemia are all immediate, reversible, treatable causes of provoked seizures. Correcting the electrolyte abnormality eliminates the need for an AED. Serum prolactin has limited diagnostic utility and a narrow sampling window. Toxicology is important but addresses a different (and less universally prevalent) precipitant. CT brain is indicated but does not need to precede the blood tests — results are often available while awaiting imaging. In a young woman with a first seizure, the electrolyte and metabolic panel is the highest-yield acute investigation after glucose.