Page 7 of 27
IM22.4 | Drug Overdose — SDL Guide
Learning Objectives
- Enumerate the commonly observed drug overdose agents in India and describe their toxicology
- Describe the clinical features, prognosis, and approach to therapy for paracetamol, benzodiazepine, tricyclic antidepressant, opioid, and salicylate overdose
- Apply the Rumack-Matthew nomogram to determine the need for N-acetylcysteine in paracetamol overdose
- Recognise the ECG features of tricyclic antidepressant toxicity and describe the role of sodium bicarbonate
- Describe the principles of enhanced elimination in drug overdose
INSTRUCTIONS
Drug overdose is the most frequent form of deliberate self-poisoning in urban India. This module covers the five most clinically important drug overdose scenarios: paracetamol, benzodiazepines, tricyclic antidepressants, opioids, and salicylates — each with a distinct mechanism, clinical signature, and management strategy.
References
- Harrison's Principles of Internal Medicine, 21st ed., Ch. 454 — Poisoning and Drug Overdose (textbook)
- API Textbook of Medicine, 10th ed., Ch. 26 — Poisoning (textbook)
- Davidson's Principles and Practice of Medicine, 24th ed., Ch. 9 — Poisoning (textbook)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A 19-year-old university student is brought to casualty at 11 pm by her roommate. She took 20 tablets of something from her bag after a heated argument with her boyfriend two hours ago. She is fully conscious, denies any symptoms, and wants to go home. Her roommate found the empty blister pack — it is a paracetamol 500 mg strip. By all external appearances, the patient looks entirely well. Her pulse is 84, BP 118/76, no jaundice, no pain. You are under pressure because the waiting room is full. But if you discharge this young woman now — if you fail to check the 4-hour paracetamol level and plot it on the Rumack-Matthew nomogram — she will present three days later in fulminant hepatic failure with coagulopathy, encephalopathy, and a serum ALT of 8,000 IU/L. This is the paradox of paracetamol overdose: the most dangerous phase is the silent period when the patient appears and feels completely well, while NAPQI is accumulating in her liver. Drug overdose does not always announce itself with drama. The clinician must know which drugs are deceptively benign in the immediate post-ingestion period and which require urgent intervention before symptoms develop.
WHY THIS MATTERS
Drug overdose is the most common form of deliberate self-poisoning in urban India, accounting for the majority of intentional poisoning presentations in tertiary care hospital emergency departments. The drugs most commonly involved reflect the local dispensing pattern: paracetamol (widely available over-the-counter, often taken in large quantities in impulsive overdoses), benzodiazepines (widely prescribed for sleep and anxiety), tricyclic antidepressants (amitriptyline and imipramine remain first-choice antidepressants in many Indian districts), tramadol and codeine (available with variable regulation), and aspirin/salicylates. Understanding drug overdose is also directly relevant to your clinical pharmacology knowledge — the toxic mechanisms of these drugs are extensions of their therapeutic mechanisms, pushed beyond the therapeutic window. NMC competency IM22.4 requires KH-level ability to enumerate, describe, and apply management principles to these specific overdose agents.
RECALL
Before proceeding, activate your pharmacology knowledge for each drug class. Paracetamol (acetaminophen) is normally metabolised primarily by glucuronidation and sulphation; a small fraction via CYP2E1 generates NAPQI (N-acetyl-p-benzoquinone imine), which is detoxified by hepatic glutathione. Benzodiazepines enhance GABA-A receptor-mediated chloride influx, producing CNS depression. Tricyclic antidepressants (TCAs) block noradrenaline and serotonin reuptake transporters (their therapeutic effect) but also block fast cardiac sodium channels (Na+ channel blockade → QRS widening → arrhythmia) and muscarinic receptors (anticholinergic features). Opioids activate Mu (μ) receptors, producing analgesia, sedation, and respiratory depression. Salicylates (aspirin) inhibit cyclooxygenase; in overdose they directly stimulate the medullary respiratory centre (producing respiratory alkalosis) and uncouple oxidative phosphorylation (producing metabolic acidosis) — a mixed acid-base picture. These mechanisms predict the clinical features and drive the antidote logic.
Paracetamol Overdose: Clinical Features and the Rumack-Matthew Nomogram
Paracetamol overdose is unique among drug poisonings in that the initial clinical presentation is deceiving — the patient appears well while a time-dependent hepatotoxic process is progressing silently. The entire management strategy is predicated on this understanding: the clinical picture at presentation tells you almost nothing about the severity of hepatic injury that will develop over the subsequent 72 hours, and serum paracetamol level plotted against the Rumack-Matthew nomogram is the only tool that allows you to predict which patients will develop liver failure and who needs N-acetylcysteine. Paracetamol is the most commonly used analgesic and antipyretic in India, widely available without prescription, and its abundance makes it the most frequently used agent in impulsive drug overdoses — particularly in young adults and adolescents after acute psychosocial stressors. The toxic dose threshold in a healthy adult is approximately 10 g (20 tablets of 500 mg), but this threshold is substantially lower in patients taking enzyme-inducing drugs such as rifampicin (widely used in India for tuberculosis), in chronic alcohol users, and in those with nutritional deficiency or glutathione depletion from fasting or systemic illness.
The clinical phases of paracetamol toxicity unfold over 96 hours:
- Phase 1 (0–24 hours): The patient is often asymptomatic or has mild nausea, vomiting, and malaise. Liver function tests are normal. This is the silent window during which NAPQI is accumulating and glutathione is being depleted.
- Phase 2 (24–72 hours): Biochemical hepatotoxicity develops. Elevated serum transaminases (AST, ALT) — initially AST rises more steeply. Right upper quadrant pain from liver capsule distension. In severe toxicity, renal function deteriorates (paracetamol-related nephrotoxicity can occur independently of hepatic injury).
- Phase 3 (72–96 hours): Peak hepatotoxicity — transaminases may reach 10,000–100,000 IU/L in severe cases. Jaundice, coagulopathy (prolonged INR from impaired clotting factor synthesis), hypoglycaemia (impaired hepatic gluconeogenesis), and hepatic encephalopathy (asterixis, confusion → coma) develop. Acute liver failure (ALF) is defined as encephalopathy with coagulopathy (INR > 1.5) in the absence of pre-existing liver disease, occurring within 26 weeks of hepatic injury.
- Phase 4 (4–14 days): Recovery phase in survivors; hepatic regeneration. Full functional recovery in most patients who survive phase 3, because paracetamol hepatotoxicity is a centrilobular (zone 3) necrosis that preserves the hepatic architecture and allows regeneration.
Mechanism of hepatotoxicity: When hepatic glutathione is depleted (by the quantity of NAPQI generated from a large paracetamol dose), NAPQI covalently binds to hepatocellular proteins, causing oxidative stress, mitochondrial dysfunction, and centrilobular hepatocyte necrosis. Factors that increase CYP2E1 activity — chronic alcohol use, fasting/malnutrition (which deplete glutathione stores), enzyme-inducing drugs (rifampicin, carbamazepine, phenytoin, isoniazid) — dramatically lower the toxic threshold dose, meaning a dose of only 5–7.5 g (10–15 tablets of 500 mg) may cause severe hepatotoxicity in these patients, compared to a threshold of approximately 10 g or more in healthy adults.
The Rumack-Matthew nomogram: A serum paracetamol level measured at 4 hours post-ingestion (the earliest reliable time point, after absorption is complete) is plotted on the Rumack-Matthew nomogram against time. If the level falls above the treatment line (approximately 150 mg/L at 4 hours, declining to ~50 mg/L at 8 hours), N-acetylcysteine (NAC) must be started immediately. The nomogram should not be used for staggered overdose (repeated smaller doses over time), modified-release preparations, or when the time of ingestion is unknown — in these scenarios, treat empirically with NAC.
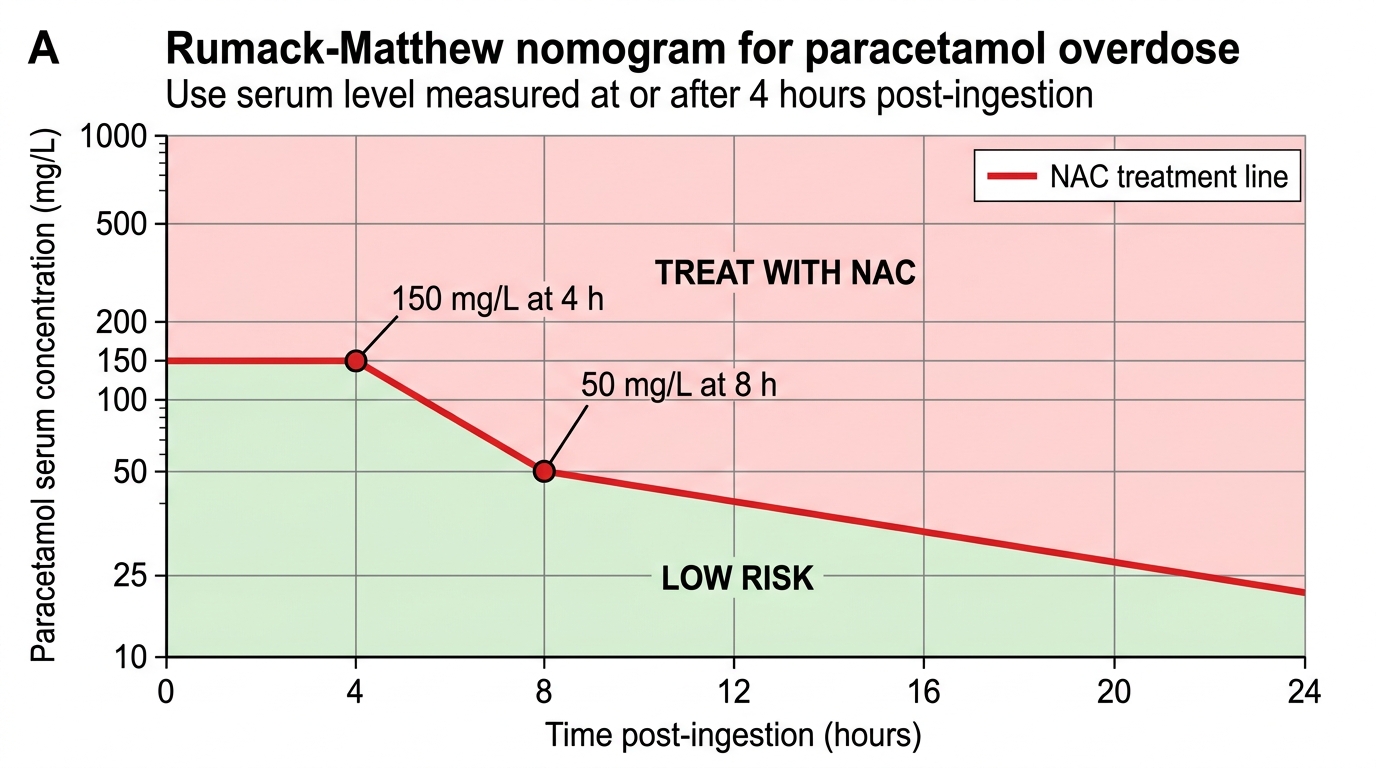
Rumack-Matthew Nomogram for Paracetamol Poisoning
Pathophysiology of Major Drug Overdose Agents
Understanding the mechanism by which each drug causes toxicity in overdose is the foundation of rational management. The toxic mechanisms of most drugs are extensions of their therapeutic mechanisms, amplified beyond the therapeutic window. For each drug discussed below, the mechanism directly predicts the dominant clinical syndrome, the most dangerous complication, and the rationale for the specific antidote or intervention.
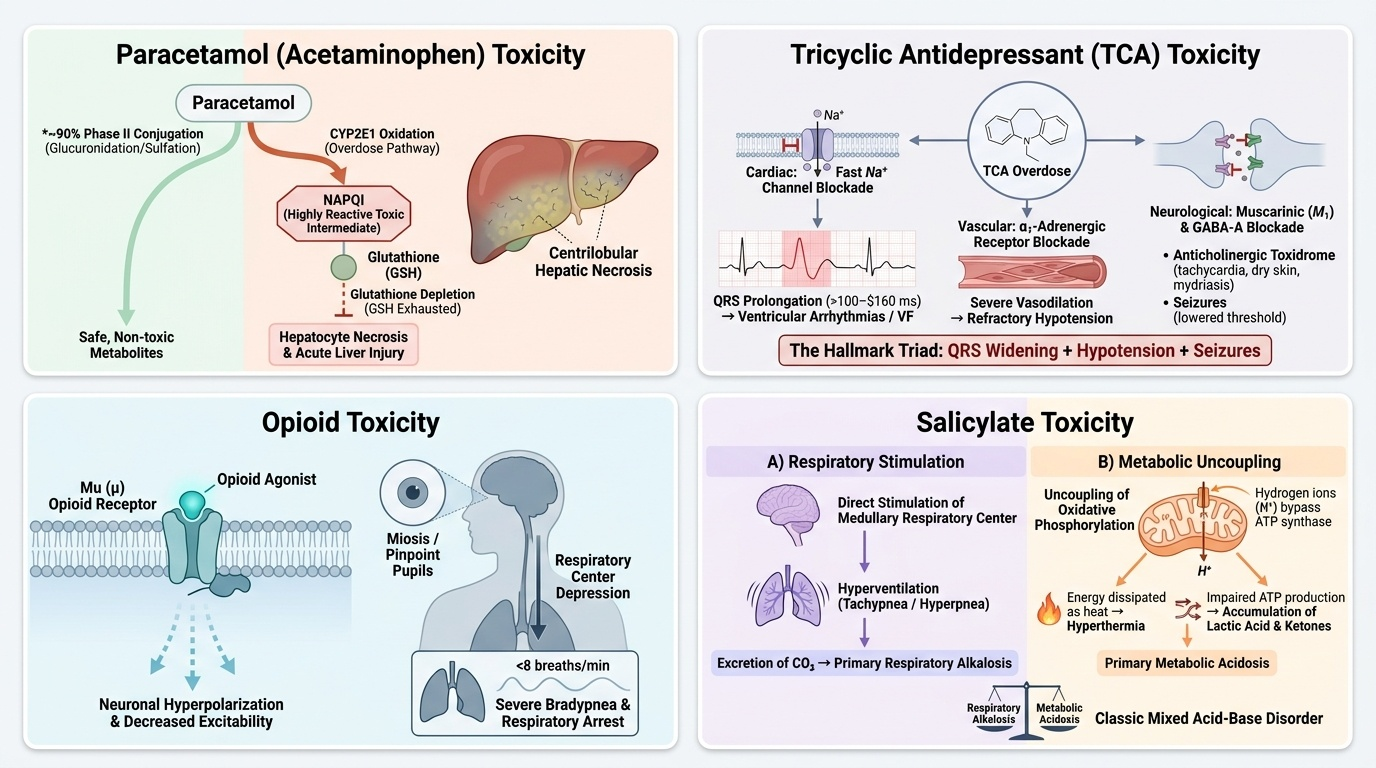
Provided image
Benzodiazepine overdose: Benzodiazepines are positive allosteric modulators of the GABA-A receptor — they increase the frequency of chloride channel opening in response to GABA, hyperpolarising the neuronal membrane and producing CNS depression. In overdose, this mechanism is simply extended: excessive chloride influx causes dose-dependent sedation, amnesia, ataxia, and finally respiratory depression. Key features: progressive sedation to coma, respiratory depression (the lethal mechanism), near-normal pupils (this distinguishes benzodiazepine overdose from opioid overdose, where pupils are pinpoint). Benzodiazepines alone rarely cause fatal respiratory failure in healthy adults — the danger is greatly increased when combined with alcohol, opioids, or other CNS depressants. The antidote is flumazenil, a competitive GABA-A antagonist; dose 0.2 mg IV, repeatable; caution in chronic BZD users (precipitates withdrawal seizures) and if TCAs co-ingested.
Tricyclic antidepressant (TCA) overdose: TCAs (amitriptyline, imipramine, clomipramine) are highly dangerous in overdose because they have three distinct and often simultaneous mechanisms of toxicity:
1. Fast Na⁺ channel blockade (quinidine-like effect): Inhibition of phase 0 depolarisation in ventricular myocardium → QRS widening → ventricular tachycardia, VF. QRS > 100 ms strongly predicts arrhythmia; QRS > 160 ms predicts VF.
2. Muscarinic receptor blockade (anticholinergic effect): Tachycardia, dry flushed skin, mydriasis, urinary retention, ileus, and delirium.
3. Alpha-1 receptor blockade: Vasodilation → hypotension — often the most clinically prominent cardiovascular effect.
4. GABA-A receptor antagonism: Seizures (distinct from benzodiazepine toxidrome — TCAs lower seizure threshold rather than raising it).
The combination of hypotension, widened QRS, and seizures is the hallmark of life-threatening TCA toxicity. Treatment: sodium bicarbonate (50–100 mEq IV bolus) is the cornerstone antidote for TCA-induced QRS widening and arrhythmia — alkalinisation reduces the free (ionised) fraction of TCA available to bind sodium channels; the extra sodium ion also competes with TCA for channel binding. Benzodiazepines for seizures. Avoid flumazenil (if patient is sedated, TCA seizures may be suppressed by endogenous BZDs — flumazenil unmasks them). Avoid physostigmine (increases risk of bradyarrhythmia and asystole in TCA toxicity).
Opioid overdose: Opioids activate μ-opioid receptors in the brainstem respiratory centres, producing dose-dependent respiratory depression — the mechanism of death. Other receptor-mediated effects: CNS depression (sedation → coma), miosis (the most specific sign — pontine μ-receptor stimulation constricts the pupil even in deep coma), reduced GI motility, urinary retention. In India, the most common opioid overdoses involve tramadol, pentazocine, and codeine-containing cough syrups. Tramadol additionally inhibits serotonin and noradrenaline reuptake and can cause seizures (unlike pure opioids). The antidote is naloxone, a pure competitive μ-receptor antagonist.
Salicylate (aspirin) overdose: The toxicology of salicylate overdose is uniquely complex because it produces a mixed acid-base disturbance through two concurrent mechanisms: (1) direct stimulation of the medullary respiratory centre by salicylate → hyperventilation → respiratory alkalosis (early and prominent); (2) uncoupling of oxidative phosphorylation in mitochondria → accumulation of organic acids (lactic acid, pyruvic acid, acetoacetic acid) → high anion gap metabolic acidosis (develops later and dominates in severe toxicity). The mixed picture — respiratory alkalosis + metabolic acidosis — is classic for salicylate. Clinical features: tinnitus and hearing loss (early, diagnostically useful), nausea and vomiting, hyperthermia (uncoupled oxidative phosphorylation generates heat), agitation, lethargy, and in severe poisoning, pulmonary oedema and coma. Hypo- or hyperglycaemia can occur (CNS glucose consumption increases disproportionately). Toxicity is worsened by acidaemia — salicylate is a weak acid (pKa 3.5), and at low pH more of it exists in the non-ionised form, which crosses the blood-brain barrier readily, worsening CNS toxicity.
SELF-CHECK
A 22-year-old woman presents 5 hours after taking 30 tablets of paracetamol 500 mg. She is asymptomatic. Serum paracetamol level at 5 hours post-ingestion is 120 mg/L, which plots BELOW the Rumack-Matthew treatment line. Liver function tests are normal. Which management is most appropriate?
A. Discharge immediately — level is below treatment line so no risk
B. Admit for 24-hour observation with repeat LFTs and INR at 24 hours
C. Start IV N-acetylcysteine immediately regardless of nomogram result
D. Give activated charcoal 50 g and repeat paracetamol level in 2 hours
Reveal Answer
Answer: B. Admit for 24-hour observation with repeat LFTs and INR at 24 hours
A paracetamol level below the treatment line at 5 hours does not mandate NAC but does not permit immediate discharge. The level should be rechecked at a later time if the ingestion timing is uncertain, and LFTs and INR should be checked at 24 hours to confirm no developing hepatotoxicity. A level below the line on a single sample is reassuring but the case history (30 tablets — 15g total in a 22-year-old) warrants observation, psychiatric assessment, and repeat biochemistry. Immediate discharge without follow-up bloods is unsafe. Activated charcoal at 5 hours is unlikely to benefit (absorption is complete by 1-2 hours post-ingestion for standard release).
Diagnosis and Investigation of Drug Overdose
The diagnosis of drug overdose is primarily clinical, supported by targeted investigations. The clinical approach has been described in the initial stabilisation and toxidrome SDL; the focus here is on the drug-specific investigations and thresholds that guide treatment decisions for each overdose agent.
Paracetamol: The definitive investigation is a serum paracetamol level at 4 hours post-ingestion (or as soon as possible after 4 hours). Earlier levels are not useful for nomogram plotting because absorption may not be complete. Plot the result on the Rumack-Matthew nomogram. If the ingestion time is unknown, use the worst-case scenario: assume the ingestion occurred at the most recent possible time that makes the level highest on the nomogram. Additionally: serum AST and ALT (may be normal in the first 24 hours even with significant overdose; any elevation at 24 hours is significant), INR (sensitive marker of hepatic synthetic function — an early rise in INR is the first sign of impending liver failure), serum creatinine (renal tubular injury can occur independently of hepatic damage), blood glucose (hypoglycaemia in hepatic failure).
Benzodiazepine: Diagnosis is clinical (sedative-hypnotic toxidrome + history). Urine toxicology screen may confirm BZD use but is not required for management decisions. ABG: respiratory acidosis (hypercapnia from hypoventilation) in significant overdose. Blood glucose (always). ECG (normal; if QRS widened, consider co-ingested TCA).
Tricyclic antidepressant: The 12-lead ECG is the most important investigation in suspected TCA toxicity. Key findings: sinus tachycardia (early and universal from anticholinergic effect), QRS duration >100 ms (predictor of ventricular arrhythmia), QTc prolongation, R wave >3 mm in aVR (a specific marker of TCA sodium channel blockade — the terminal 40 ms frontal plane axis shifts rightward due to slowed conduction in the right ventricle, producing a positive deflection in the rightward lead aVR). Serial ECGs every 15–30 minutes in significant overdose. Serum TCA levels are not routinely used to guide management — the ECG findings are the clinical decision driver. ABG: respiratory alkalosis early (hyperventilation); acidosis worsens sodium channel blockade (a dangerous positive feedback loop justifying bicarbonate therapy).
Opioid: Diagnosis is clinical (opioid toxidrome: miosis + respiratory depression + coma). ABG: respiratory acidosis (hypercapnia, hypoxia). Blood glucose. Urine toxicology if available. Naloxone response is both diagnostic and therapeutic.
Salicylate: Serum salicylate level is the key investigation. The Done nomogram (for salicylate) is less reliable than the Rumack-Matthew for paracetamol; clinical severity grading (mild: salicylate 30–50 mg/dL; moderate: 50–100 mg/dL; severe: >100 mg/dL) correlates roughly with clinical features but serial clinical assessment is more important. ABG: the classic mixed picture of respiratory alkalosis (low PaCO₂, high pH early) + high anion gap metabolic acidosis (low HCO₃ dominating later). Serum electrolytes (anion gap calculation). Blood glucose (hypoglycaemia especially in children). Urine pH (a bedside marker of urinary alkalinisation efficacy — target urine pH 7.5–8.5 for enhanced elimination).
⚑ AI image — pending faculty review (auto-QA score 7/10; best of 3 attempts)
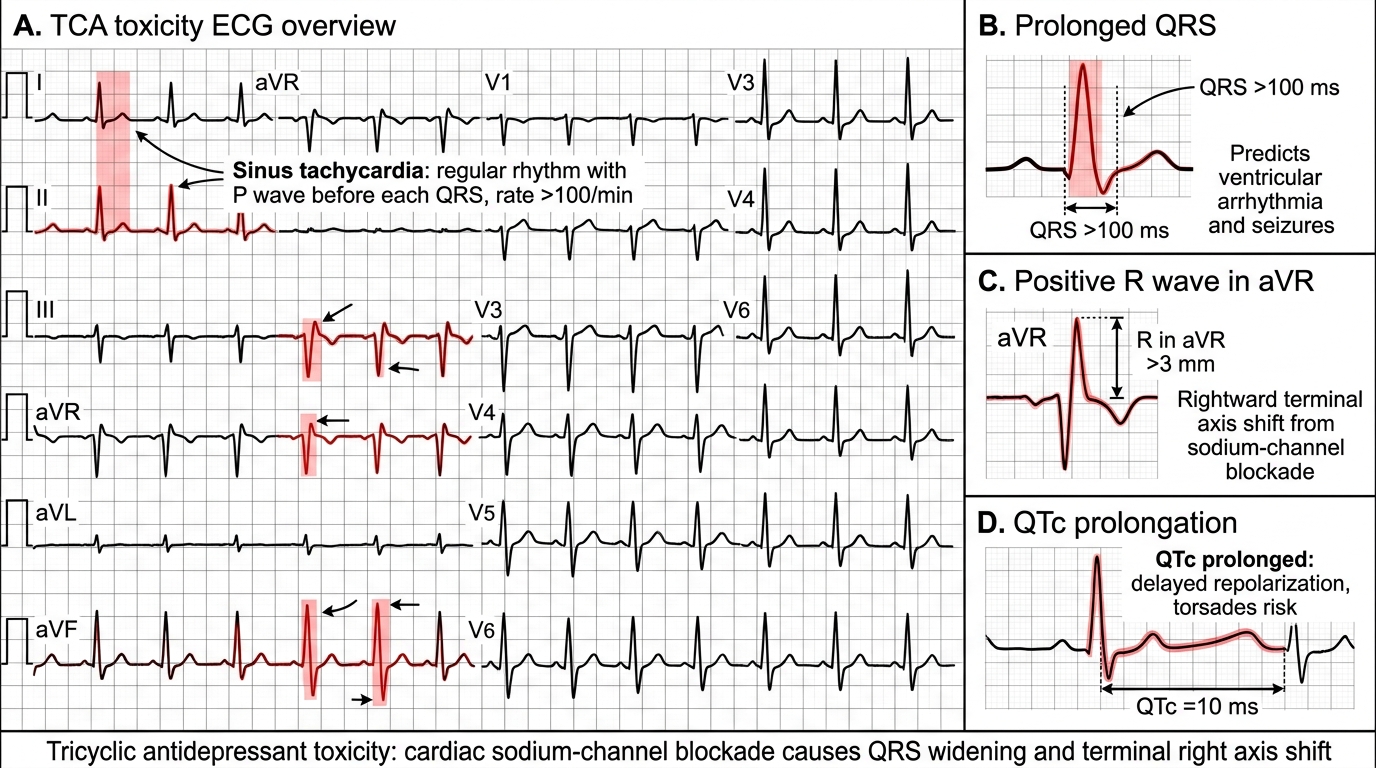
ECG Features of TCA Toxicity