Page 10 of 27
IM22.6 | Insecticide Poisoning — SDL Guide
Learning Objectives
- Describe the toxicology of organophosphate and carbamate insecticides including their mechanism of acetylcholinesterase inhibition
- Enumerate the clinical features of organophosphate and carbamate poisoning using the cholinergic toxidrome framework
- Describe the specific complications including intermediate syndrome and organophosphate-induced delayed neuropathy
- Outline the management of organophosphate poisoning including atropine dosing protocol and pralidoxime indications
- Distinguish organophosphate from carbamate poisoning in clinical and management terms
INSTRUCTIONS
Organophosphate and carbamate insecticide poisoning is the most common cause of acute cholinergic crisis in India, with high mortality from respiratory failure. This module covers the biochemistry, clinical syndrome, complications, and management — with particular emphasis on the correct atropine dosing protocol, which is the single most important determinant of survival.
References
- Harrison's Principles of Internal Medicine, 21st ed., Ch. 454 — Poisoning and Drug Overdose (textbook)
- API Textbook of Medicine, 10th ed., Ch. 26 — Poisoning (textbook)
- Davidson's Principles and Practice of Medicine, 24th ed., Ch. 9 — Poisoning (textbook)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A 40-year-old farmer is admitted to your district hospital in a vegetative-looking state. His son says he collapsed in the field four hours ago and was brought by auto-rickshaw. On examination: GCS 7, temperature 36.4°C, blood pressure 70/40 mmHg, pulse 38 bpm, respiratory rate 6/min with audible bubbling in the chest, pupils pinpoint at 1 mm, profuse salivation pooling under his tongue, muscle fasciculations visible across his chest wall. Your junior colleague reaches for the first atropine ampoule — 0.6 mg — and gives it intravenously. The pulse rises briefly to 52. He gives a second 0.6 mg. Nothing else happens. The secretions continue. The wheezing worsens. The patient is transferred to your ICU — still secreting, still hypotensive, still dying. Three hours later, having received a total of 6 mg of atropine over 3 hours, the patient is intubated and on a ventilator. The cause of death the next morning: refractory bronchospasm and secretion aspiration. The atropine was given. But it was not given correctly. The first hour of atropinisation — when it matters most — was spent in hesitation, small doses, and waiting for pupil dilation that never needed to come. Understanding the correct atropine protocol for organophosphate poisoning is, quite literally, the difference between life and death.
WHY THIS MATTERS
Organophosphate (OP) and carbamate insecticide poisoning is the most common cause of pesticide poisoning in India, with tens of thousands of cases annually concentrated in agricultural states (Andhra Pradesh, Telangana, Punjab, Maharashtra, Tamil Nadu). It is also the most common cause of the acute cholinergic toxidrome and the most important context for atropine therapy in emergency medicine. The case fatality rate in district hospital settings is high — primarily because of inadequate atropine dosing, delayed recognition of intermediate syndrome, and insufficient ventilatory support infrastructure. As a final-year student entering internship and rural postings, you will almost certainly encounter this emergency. NMC competency IM22.6 requires KH-level ability to describe and discuss the toxicology, clinical features, complications, prognosis, and specific management of OP and carbamate poisoning — not just know it, but be able to apply it.
RECALL
From your biochemistry and pharmacology studies, recall the cholinergic synapse: the nerve terminal releases acetylcholine (ACh) into the synaptic cleft; ACh binds to postsynaptic receptors; acetylcholinesterase (AChE) in the cleft hydrolyses ACh to choline and acetate, terminating the signal. Recall the two major receptor types: muscarinic receptors (M1–M5) at postganglionic parasympathetic nerve endings — heart (bradycardia), smooth muscle (bronchospasm, GI motility), exocrine glands (secretions), pupil (miosis); and nicotinic receptors at the neuromuscular junction (NMJ) and autonomic ganglia — stimulation causes fasciculations, progressing to depolarisation block and paralysis. From pharmacology, recall that atropine is a competitive antagonist at muscarinic receptors (no effect on nicotinic), and that pralidoxime (2-PAM) is an oxime that can reactivate acetylcholinesterase if the enzyme has not yet undergone irreversible ageing.
Clinical Presentation of Organophosphate and Carbamate Poisoning
Organophosphate and carbamate poisoning presents as the acute cholinergic toxidrome — a syndrome that is unmistakable once recognised but can be confused with other conditions (sepsis, brainstem stroke, opioid overdose) if the examiner is not looking for the complete picture. The clinical features arise from excess acetylcholine accumulation at three anatomical sites, each producing a distinct sub-syndrome that must be assessed separately: (1) muscarinic receptors at postganglionic parasympathetic nerve endings; (2) nicotinic receptors at the neuromuscular junction; and (3) nicotinic/muscarinic receptors in the central nervous system. In a severe case, all three sub-syndromes are present simultaneously.
Route of exposure determines the onset speed and initial dominant symptoms. Dermal exposure (most common in agricultural settings — spraying without personal protective equipment) typically produces a slower onset over 30 minutes to several hours, with skin and eye symptoms (miosis, lacrimation) preceding systemic features. Oral ingestion (deliberate self-harm or accidental — most dangerous) produces rapid systemic absorption and presentation within 30–60 minutes. Inhalational exposure (fumigation accidents) causes rapid CNS and pulmonary effects. The history of exposure — working with pesticides, smell of organophosphate on clothing, finding an agricultural bottle — is critical.
Muscarinic features (SLUDGE/DUMBELS): The classic mnemonic encompasses all the exocrine and smooth muscle effects of parasympathetic overactivity: Salivation (drooling, pooling of saliva), Lacrimation (tearing), Urination (incontinence), Defecation (faecal incontinence, diarrhoea), GI distress (nausea, vomiting, cramping), Emesis. Additional muscarinic features: miosis (pinpoint pupils — bilateral, reactive or near-fixed), bradycardia (often severe — 20–40 bpm), bronchospasm and bronchorrhoea (the life-threatening features — excess secretions in the airways cause asphyxiation), hypotension (both from bradycardia and direct vasodilation), and diaphoresis (profuse sweating). The combination of miosis + bradycardia + bronchorrhoea in a profusely sweating patient is pathognomonic.
Nicotinic features (NMJ): Fasciculations — involuntary, visible, rapid twitching of individual muscle groups — are the hallmark nicotinic sign and are virtually diagnostic of organophosphate poisoning in context. Initially, nicotinic stimulation at the NMJ causes fasciculations; as acetylcholine accumulates further, sustained depolarisation block develops — leading to flaccid muscle weakness and eventually paralysis. The respiratory muscles are especially vulnerable: diaphragmatic paralysis is a major cause of respiratory failure, superimposed on the bronchospasm and bronchorrhoea already described. Nicotinic stimulation at sympathetic ganglia causes an initial phase of tachycardia and hypertension that may precede or temporarily mask the muscarinic bradycardia — this paradoxical tachycardia in the early phase can cause diagnostic confusion.
CNS features: Anxiety, restlessness, and ataxia are early; headache, dizziness, and confusion follow; seizures (common in severe poisoning — both from direct CNS effects and from hypoxia); and finally coma. Seizures in OP poisoning are muscarinic-independent (benzodiazepines are the treatment of choice; atropine does not prevent or treat seizures in OP poisoning).
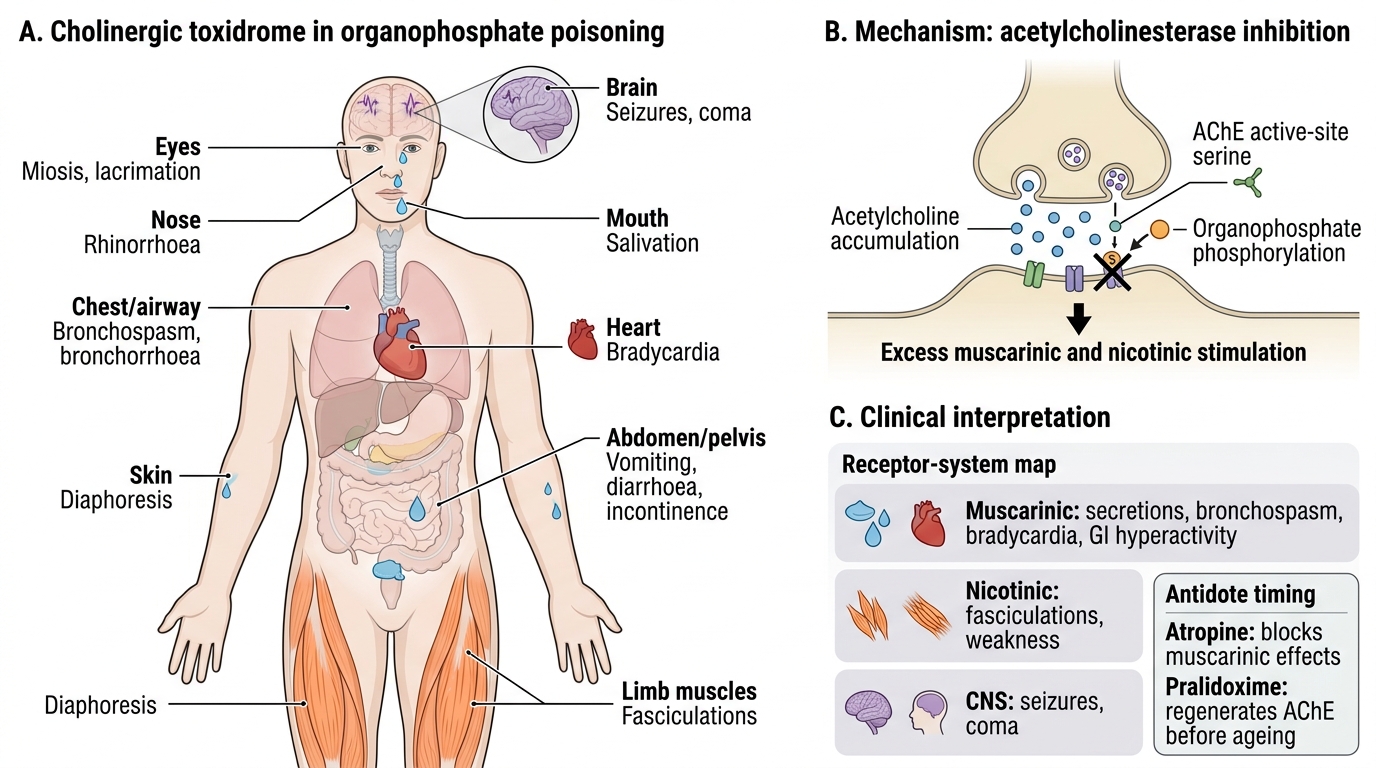
Cholinergic Toxidrome in Organophosphate Poisoning
Toxicology and Pathophysiology of Acetylcholinesterase Inhibition
The toxicology of organophosphate and carbamate compounds rests on a single biochemical mechanism: inhibition of acetylcholinesterase (AChE), the enzyme responsible for hydrolysing and inactivating acetylcholine at cholinergic synapses. Understanding the molecular mechanism — and the critical difference between organophosphate and carbamate inhibition — explains why pralidoxime works for one and not the other, and why timing of antidote administration is so crucial.
AChE inhibition mechanism:
Acetylcholinesterase functions by a serine hydrolase mechanism: the active site serine residue binds ACh, hydrolyses the ester bond, and is rapidly regenerated. Organophosphates irreversibly phosphorylate the active site serine via a covalent bond, forming a phosphorylated AChE complex. This phosphorylation is initially reversible — oximes like pralidoxime can displace the phosphate group and regenerate active AChE — but it undergoes ageing (strengthening of the covalent bond) over time, making it truly irreversible. Ageing time varies: hours for most common agricultural OPs (chlorpyrifos, malathion, profenofos, monocrotophos, dimethoate); but only minutes for military nerve agents (soman — not relevant in India but pharmacologically important). Carbamates reversibly carbamylate the serine residue — the carbamylation spontaneously reverses within hours without any pharmacological intervention, which is why pralidoxime is not required for carbamate poisoning.
Consequences of AChE inhibition:
With AChE blocked, every cholinergic nerve impulse causes acetylcholine accumulation in the synapse. The consequence at each receptor type has been described above. Serum cholinesterase activity (RBC and plasma AChE) is a useful diagnostic and severity marker: normal RBC AChE 6.9–10.3 U/mL; activity < 50% of normal confirms cholinergic poisoning; < 20% indicates severe poisoning requiring intensive management. Note, however, that serum cholinesterase levels lag behind the clinical picture — clinical assessment drives treatment; the level confirms and guides severity stratification.
Distinguishing OP from carbamate:
- Organophosphate: irreversible AChE inhibition (ageing); severe and prolonged toxicity; serum cholinesterase suppressed for days to weeks; pralidoxime INDICATED (before ageing); intermediate syndrome possible (see below).
- Carbamate: reversible, spontaneously reversible carbamylation; clinically similar but shorter duration (hours); serum cholinesterase recovers quickly; pralidoxime NOT required (carbamylation reverses spontaneously; pralidoxime may worsen toxicity in some studies); atropine alone is sufficient.
Common organophosphate agents in India: chlorpyrifos, malathion, quinalphos, profenofos, monocrotophos, dimethoate, dichlorvos (DDVP). Highly toxic agents: mevinphos, parathion. Common carbamates: carbofuran, methomyl, aldicarb.
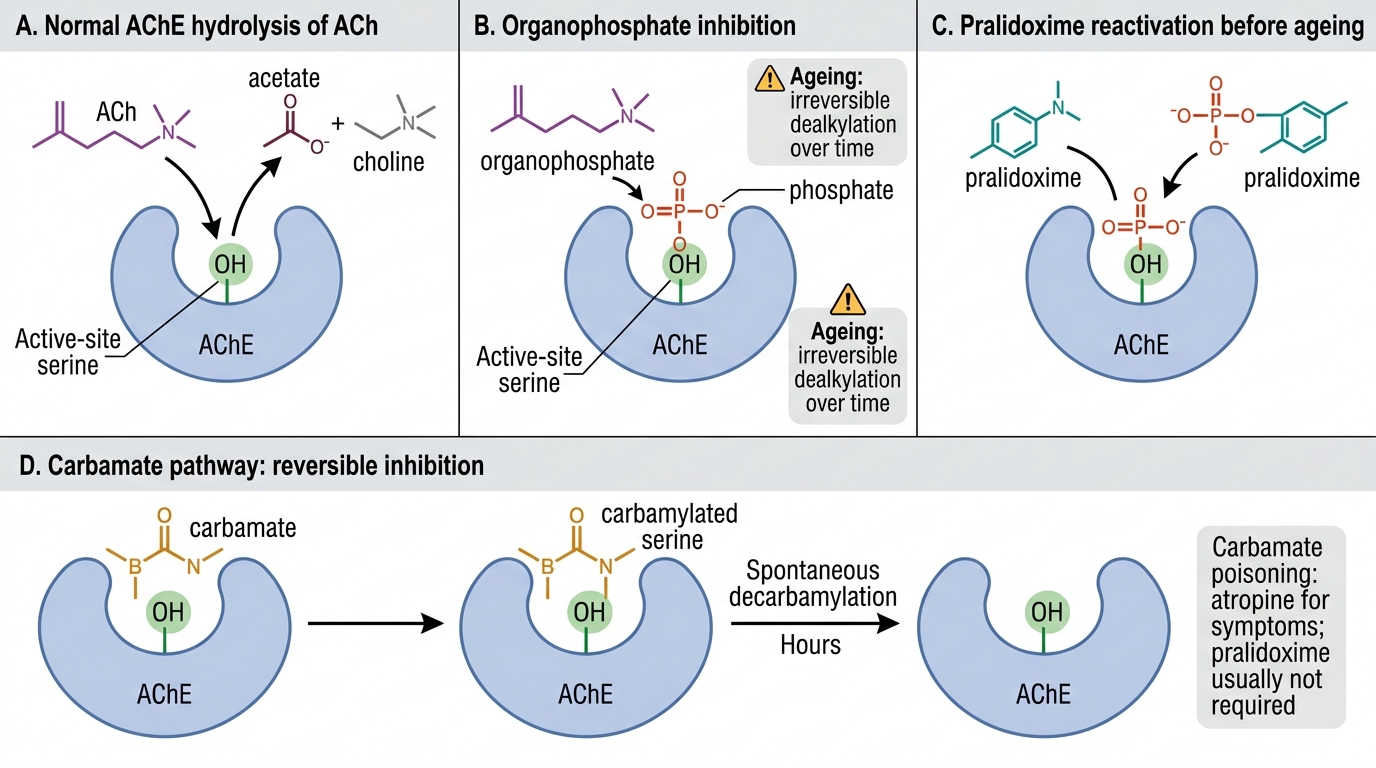
AChE Inhibition and Reactivation in Poisoning
SELF-CHECK
A 32-year-old farmer presents with acute cholinergic toxidrome after spraying carbofuran insecticide without protective equipment. He has profuse salivation, miosis, bronchospasm, and bradycardia. Which statement about his management is MOST accurate?
A. Atropine and pralidoxime are both required as in organophosphate poisoning
B. Pralidoxime is NOT required as carbamate inhibition of AChE reverses spontaneously; atropine alone is sufficient
C. Pralidoxime must be given within 1 hour before carbamylation becomes irreversible
D. Atropine is contraindicated in carbamate poisoning due to risk of anticholinergic crisis
Reveal Answer
Answer: B. Pralidoxime is NOT required as carbamate inhibition of AChE reverses spontaneously; atropine alone is sufficient
Carbamates reversibly carbamylate the AChE active site serine — the carbamylation spontaneously reverses within hours without any antidote. Therefore pralidoxime is NOT required for carbamate poisoning; atropine alone (to block the excess acetylcholine effect at muscarinic receptors) is sufficient. This distinguishes carbamate from organophosphate poisoning: in OPs, pralidoxime is needed because the phosphorylation undergoes irreversible ageing. Atropine is certainly indicated in carbamate poisoning (it reverses the muscarinic features) and is never contraindicated.
Diagnosis and Severity Assessment
Diagnosis of organophosphate or carbamate poisoning is primarily clinical — the cholinergic toxidrome in the context of pesticide exposure is unmistakable when the full picture is assembled. Investigations serve three purposes: they confirm the biochemical diagnosis, quantify the severity of acetylcholinesterase inhibition, and guide the intensity of specific interventions (atropine dosing, ventilatory support, pralidoxime duration). A structured approach to severity assessment prevents the common clinical error of under-estimating the severity of an apparently alert patient and over-estimating the safety of one who has transiently improved. Critically, severity assessment is continuous — not a one-time admission score — because both clinical deterioration (towards respiratory failure) and the development of delayed complications (intermediate syndrome) require ongoing monitoring throughout the first four days of the hospital course. The following investigations are ordered in parallel with atropinisation, not as a prerequisite for it — do not delay atropine for blood results.
Clinical diagnosis: The presence of miosis + bradycardia + bronchospasm/bronchorrhoea + SLUDGE features + fasciculations in a patient with a history of agricultural chemical exposure is diagnostic. Do not wait for laboratory results to begin atropinisation. The smell of garlic or petroleum on the patient, clothing, or vomitus is a clinical clue to OP exposure.
Investigations:
- Serum cholinesterase (RBC AChE and plasma pseudocholinesterase): Normal RBC AChE 6.9–10.3 U/mL; levels fall proportionally to the degree of inhibition. Severity grading: 50–75% suppression = mild; 25–50% = moderate; < 25% = severe. Plasma pseudocholinesterase is easier to measure but more sensitive to other factors (liver disease, pregnancy, genetic variants). Serial measurements track recovery — cholinesterase activity rises as AChE is resynthesised (new enzyme — not reactivation), taking days to weeks after OP poisoning.
- ABG: Hypoxia and hypercapnia are early in significant bronchospasm and respiratory muscle involvement. Metabolic acidosis from tissue hypoperfusion in severe shock.
- 12-lead ECG: QTc prolongation is common (direct OP effect on cardiac conduction); bradycardia, AV block, ventricular arrhythmias. ECG monitoring is mandatory throughout treatment.
- CXR: Pulmonary oedema (non-cardiogenic, from bronchorrhoea and aspiration); collapse/consolidation.
- Blood glucose: Hypoglycaemia or hyperglycaemia (stress response); monitor closely.
- Serum electrolytes: Hypokalaemia is common (from vomiting, diarrhoea, and alkalosis from bicarbonate used in some protocols).
Severity grading (Peradeniya Organophosphorus Poisoning scale and WHO/WHO classification):
Severity can be graded clinically as mild (miosis, salivation, normal or mildly decreased consciousness), moderate (bronchospasm, bradycardia, diaphoresis, moderate CNS depression), or severe (coma, seizures, respiratory failure, severe bradycardia < 40 bpm, cyanosis). Severe cases require ICU admission, mechanical ventilation, and intensive atropine titration.
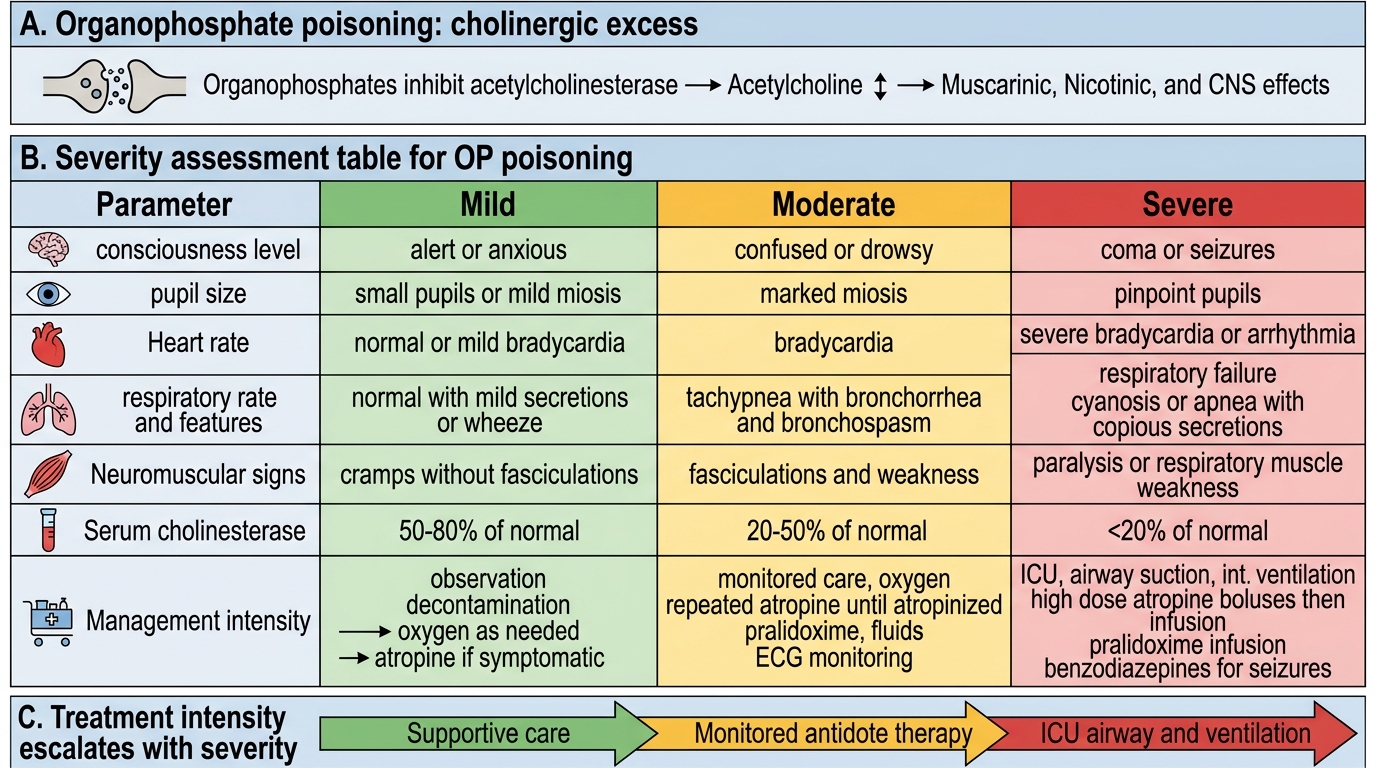
Severity Grading and Management of Organophosphate Poisoning