Page 2 of 22
IM28.1-7 | Obstructive Airway Disease Foundations — SDL Guide (Part 2)
Pathophysiology of Hypoxia and Hypercapnia
The pathophysiological consequences of obstructive airway disease ultimately manifest as disordered gas exchange — specifically hypoxia (reduced PaO₂) and, in advanced disease, hypercapnia (elevated PaCO₂). Understanding the mechanisms by which airway obstruction produces these gas exchange defects is essential for interpreting arterial blood gas results, deciding when to initiate oxygen therapy, and understanding why supplemental oxygen must be used cautiously in hypercapnic COPD patients — one of the most important and frequently tested clinical principles in respiratory medicine.
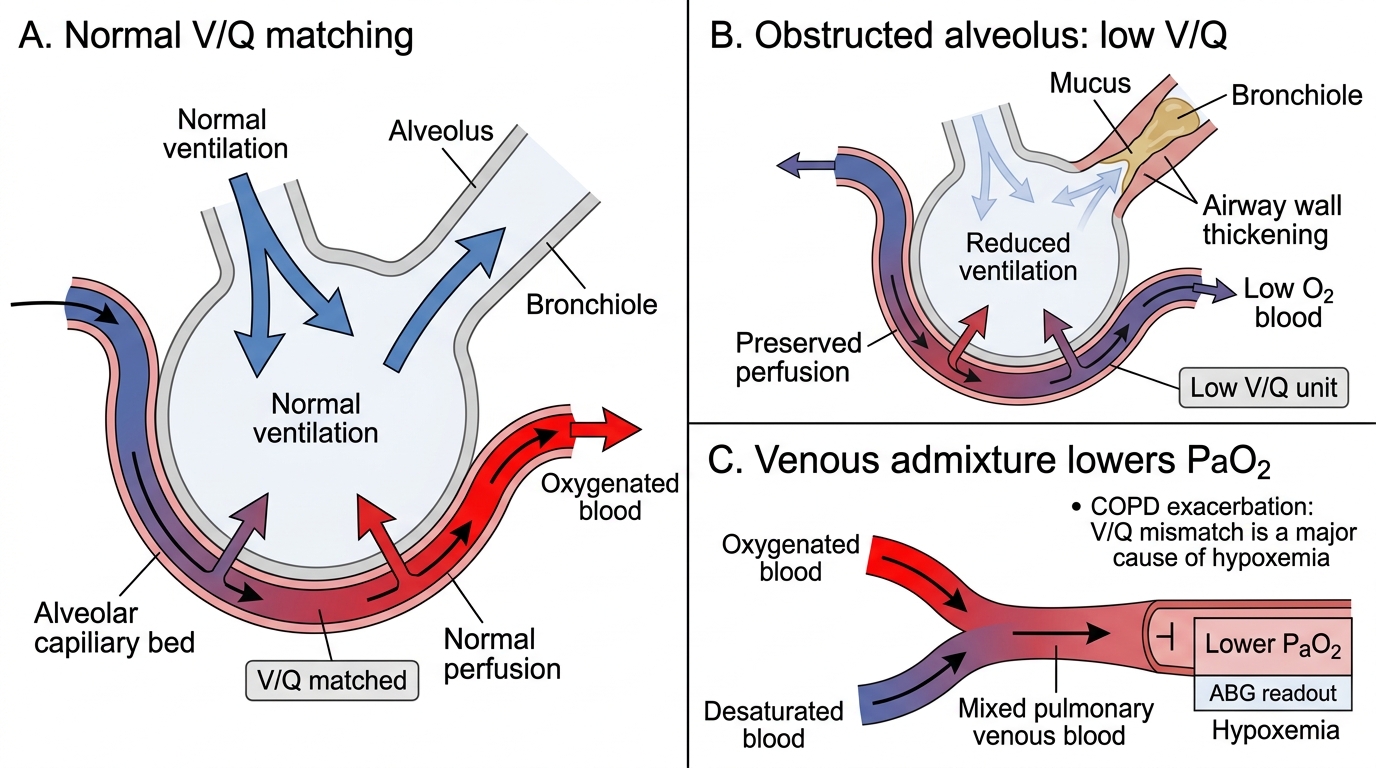
Normal gas exchange depends on the matching of ventilation (V) and perfusion (Q) at the alveolar level. In health, the V/Q ratio is approximately 0.8–1.0 throughout most of the lung, ensuring efficient transfer of O₂ into the blood and CO₂ out. Airway obstruction disrupts this matching — a narrowed or collapsed airway reduces ventilation to the alveoli it supplies while blood flow continues, creating regions of low V/Q where blood passes through poorly-ventilated alveoli and emerges with reduced O₂ saturation and elevated CO₂. This V/Q mismatch is the predominant mechanism of hypoxia in both asthma and COPD.
Hypoxia mechanisms in obstructive airway disease:
1. V/Q mismatch (dominant mechanism) — obstructed airways reduce regional ventilation; blood flow persists through these areas; desaturated blood mixes with blood from normally ventilated areas, reducing overall PaO₂
2. Alveolar hypoventilation — in severe obstruction or fatigue of the respiratory muscles, overall alveolar ventilation falls; both PaO₂ falls and PaCO₂ rises (as below)
3. Hypoxic pulmonary vasoconstriction (HPV) — in response to localised alveolar hypoxia, the pulmonary arterioles supplying that region constrict, diverting blood away from hypoxic areas toward better-ventilated areas; this is a compensatory mechanism that improves V/Q matching. However, in severe widespread disease, global hypoxaemia drives global pulmonary vasoconstriction, raising pulmonary artery pressure and eventually causing cor pulmonale (right ventricular hypertrophy and failure from pulmonary hypertension)
Hypercapnia mechanisms:
In mild-to-moderate obstructive disease, hypercapnia does not develop despite hypoxia, because the carotid body chemoreceptors detect the falling PaO₂ and stimulate increased ventilation — this increased ventilatory drive washes out the accumulated CO₂ and maintains PaCO₂ near normal (or even below normal, causing mild hypocapnia). However, in severe COPD with air trapping and dynamic hyperinflation, the respiratory muscles operate at a mechanical disadvantage — the diaphragm is flattened, accessory muscles are recruited, and every breath requires disproportionate effort. When the load on the respiratory muscles exceeds their capacity, alveolar hypoventilation supervenes, PaCO₂ rises, and type-2 respiratory failure (hypoxia + hypercapnia) develops. Chronic CO₂ retention leads to bicarbonate retention by the kidney, producing a compensated respiratory acidosis (elevated PaCO₂, elevated HCO₃⁻, pH near-normal).
The hypoxic drive concept and oxygen therapy in COPD: in chronic CO₂ retainers, the central respiratory centre (medullary chemoreceptors) becomes relatively insensitive to elevated CO₂ (because CSF bicarbonate rises to buffer the chronic CO₂ elevation). The remaining ventilatory drive is maintained by peripheral hypoxic drive from carotid body stimulation by the low PaO₂. This is why administering high-flow uncontrolled oxygen to a chronic hypercapnic COPD patient can cause the PaO₂ to rise rapidly, suppress the hypoxic drive, reduce ventilatory effort, lead to further CO₂ retention and a rising PaCO₂, and precipitate CO₂ narcosis and respiratory arrest. The safe approach is controlled low-flow oxygen (target SpO₂ 88–92% in known chronic CO₂ retainers, as opposed to 94–98% in most other patients) — the Venturi mask is designed precisely for this purpose, delivering precise FiO₂ of 24%, 28%, or 35% regardless of flow rate.
V/Q Mismatch in Obstructive Airway Disease
SELF-CHECK
A 65-year-old male with known COPD is brought to emergency with acute exacerbation. His ABG on room air shows pH 7.32, PaO₂ 48 mmHg, PaCO₂ 68 mmHg, HCO₃⁻ 32 mEq/L. What does this ABG pattern indicate and what oxygen target is appropriate?
A. Metabolic acidosis; target SpO₂ 94–98%
B. Acute respiratory alkalosis; oxygen therapy not indicated
C. Compensated respiratory acidosis (chronic CO₂ retention); target SpO₂ 88–92%
D. Mixed metabolic and respiratory acidosis; target SpO₂ 94–98%
Reveal Answer
Answer: C. Compensated respiratory acidosis (chronic CO₂ retention); target SpO₂ 88–92%
The ABG shows: elevated PaCO₂ (68 mmHg — hypercapnia), low pH (7.32 — acidosis), and elevated HCO₃⁻ (32 mEq/L — metabolic compensation). Elevated bicarbonate indicates chronic CO₂ retention with renal compensation — this is a compensated respiratory acidosis, indicating this patient is a chronic CO₂ retainer. The acute-on-chronic pattern is suggested by the pH of 7.32 (compensated would normalise closer to 7.38–7.40). In chronic CO₂ retainers, the safe oxygen target is SpO₂ 88–92% (not 94–98%) to avoid suppressing the residual hypoxic drive, which could worsen hypoventilation and CO₂ retention. Giving uncontrolled high-flow O₂ risks CO₂ narcosis.
Genetics of Alpha-1 Antitrypsin Deficiency and Emphysema
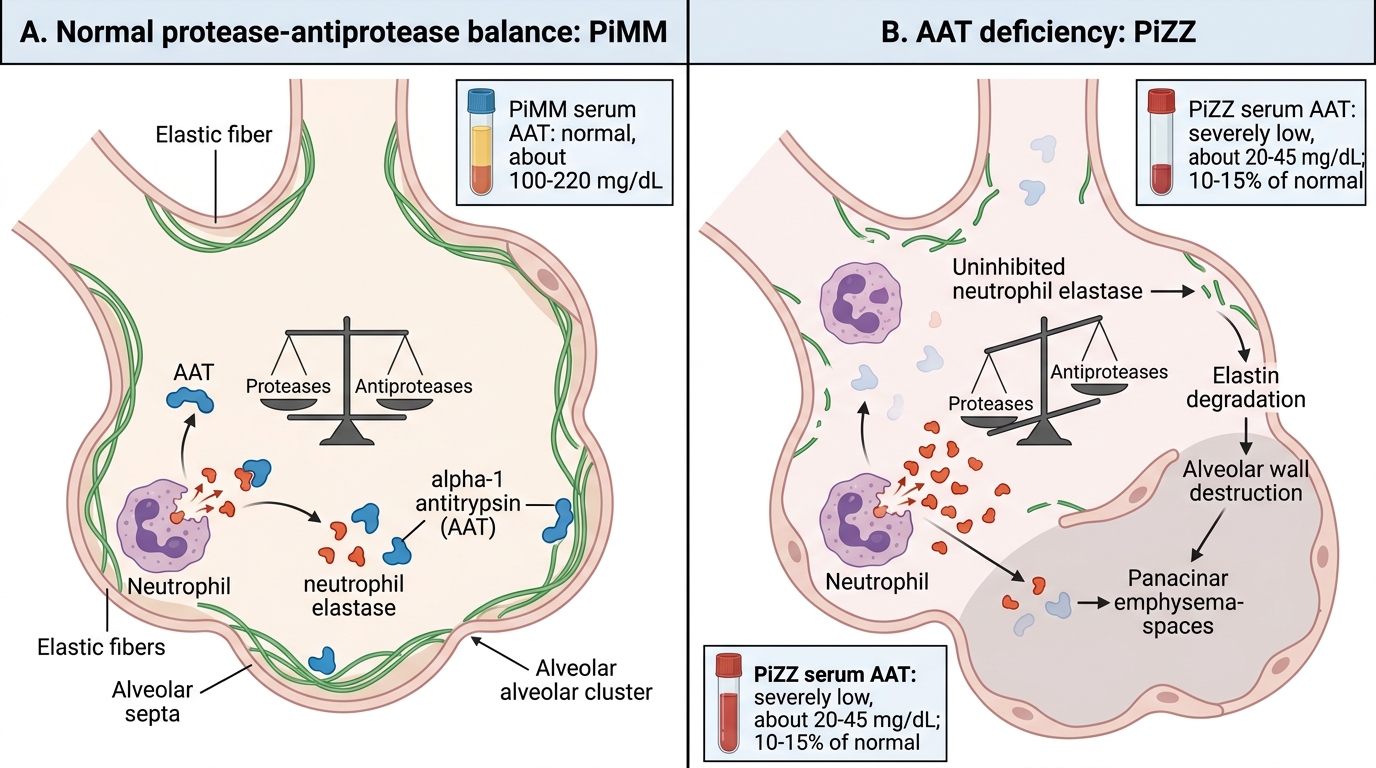
Alpha-1 antitrypsin (AAT) is the most abundant circulating serine protease inhibitor (serpin) in plasma. It is synthesised primarily in the liver and secreted into the bloodstream, from where it reaches the lungs via the alveolar fluid. Its primary function in the lung is to inhibit neutrophil elastase — an enzyme released by neutrophils during inflammatory responses that degrades the elastin scaffolding of alveolar walls. The concept that underpins emphysema pathogenesis is the protease-antiprotease hypothesis: emphysema results from an imbalance in which proteolytic enzymes (elastase, matrix metalloproteinases) overwhelm their natural inhibitors, leading to progressive alveolar wall destruction.
In normal individuals, sufficient AAT is produced to neutralise neutrophil elastase released during the lung's daily microbial encounters and particle clearance. In AAT deficiency, this protective inhibitory reservoir is absent or severely reduced, and even the normal load of neutrophil elastase from everyday lung defense is sufficient to progressively destroy alveolar walls — causing panacinar emphysema (affecting the entire acinus, predominantly in the lung bases — in contrast to the centri-acinar emphysema of tobacco-related COPD, which affects respiratory bronchioles and is upper-lobe predominant).
Genetics of AAT deficiency:
The AAT gene (SERPINA1) is located on chromosome 14q32.1. The protein is encoded by a highly polymorphic gene with over 100 alleles identified, classified by isoelectric focusing migration speed (Pi system — Protease inhibitor). The commonest clinically relevant alleles are:
- PiMM (wildtype): normal phenotype; serum AAT ~100–200 mg/dL (100% of normal); no increased emphysema risk
- PiMS and PiSS: mildly reduced AAT (60% and ~60% of normal respectively); modest clinical risk
- PiMZ (heterozygous): serum AAT ~60% of normal; modestly increased risk, especially with smoking
- PiZZ (homozygous): serum AAT <15% of normal (typically 20–50 mg/dL); classical AAT deficiency — causes early-onset panacinar emphysema (onset 40–50 years even without smoking, 30–40 years in smokers) AND predisposes to liver disease (neonatal hepatitis, cirrhosis) because misfolded Z-protein polymerises and accumulates in hepatocytes rather than being secreted. The Z allele (Glu342Lys substitution) causes the protein to misfold and polymerise in the ER
- PiNull: no AAT produced; extreme emphysema risk
Inheritance: AAT deficiency follows an autosomal co-dominant inheritance pattern — both alleles are expressed, and the phenotype of heterozygotes is intermediate between the two homozygous phenotypes. Family screening is therefore important: if a proband is PiZZ, each sibling has a 1-in-4 chance of being PiZZ if both parents carry the Z allele.
Epidemiology: PiZZ homozygosity occurs in approximately 1 in 1,700–5,000 people of European ancestry; it is less common in Asian and African populations. AAT deficiency accounts for approximately 1–2% of all COPD cases, but it is important because it represents a diagnosable, potentially treatable cause of COPD with familial implications.
Screening and augmentation therapy: all patients with COPD or emphysema diagnosed below the age of 45 years, non-smokers with emphysema, or patients with a strong family history should be tested for AAT deficiency with serum AAT level and phenotyping/genotyping. Augmentation therapy (infusion of purified pooled-plasma AAT) is approved for PiZZ patients with established emphysema; evidence for slowing lung function decline is modest but there is a signal for reduced CT-measured emphysema progression. Smoking cessation is the most important single intervention — smoking dramatically accelerates the neutrophilic lung injury and the rate of FEV1 decline.
Protease-Antiprotease Balance in Alpha-1 Antitrypsin Deficiency
Environmental Factors, Allergic and Non-Allergic Precipitants
The distinction between the causes of obstructive airway disease (the factors that establish the disease over months to years) and the precipitants of acute exacerbations (the factors that worsen control acutely, often within minutes to hours) is clinically important, though the two categories overlap. Environmental factors can both cause and precipitate disease. Identifying the specific environmental and allergic/non-allergic triggers in each patient is a central part of the individualised management plan, because avoidance and mitigation of modifiable triggers often reduces exacerbation frequency and severity significantly.
Provided image
Tobacco smoking is the dominant environmental cause of COPD worldwide, responsible for approximately 80–90% of cases in high-income countries. Cigarette smoke contains over 4,000 chemicals including reactive oxygen species (ROS), acrolein, and particulate matter that activate airway macrophages and neutrophils, drive a sustained neutrophilic-predominant inflammation, damage airway epithelial cells, impair mucociliary clearance, and activate protease pathways that degrade connective tissue. Bidi smoking is quantitatively important in India — a bidi delivers higher concentrations of tar and nicotine per puff than a cigarette, and bidi smokers develop COPD at lower pack-year exposures than cigarette smokers. Passive (second-hand) smoke exposure is an established cause of childhood asthma and contributes to COPD risk in non-smoking adults with sustained household or occupational exposure.
Biomass fuel smoke is the leading cause of COPD in non-smoking Indian women, arising from indoor burning of wood, crop residue, cow dung, and coal for cooking and heating in poorly-ventilated homes. Women in these households can have cumulative exposures to particulate matter equivalent to smoking 20–40 cigarettes per day. WHO estimates that approximately 3 billion people worldwide use solid fuels for cooking — making biomass-related lung disease a global issue. The inflammatory profile of biomass-related COPD may differ from tobacco COPD (more mixed inflammatory infiltrate, more wall-thickening and less emphysema), but the clinical and spirometric manifestations are broadly similar.
Occupational exposures: a wide range of occupational dust and chemical exposures are established causes of both occupational asthma and COPD:
- Organic dusts: flour dust (occupational asthma in bakers), cotton dust (causing byssinosis — Monday morning chest tightness in textile workers, particularly characteristic), grain dust, wood dust, laboratory animal proteins
- Inorganic dusts: coal dust (combined coal workers' pneumoconiosis and airflow obstruction), silica (silicosis with accelerated airflow obstruction), asbestos
- Chemical agents: isocyanates (the most common cause of occupational asthma in industrialised countries — in paint sprayers, polyurethane foam workers, car body repairers), reactive dyes, formaldehyde, chlorine gas, sulfur dioxide, and metal fumes
- Agricultural exposures: endotoxin-rich organic dusts (swine confinement workers, poultry workers), pesticide sprays
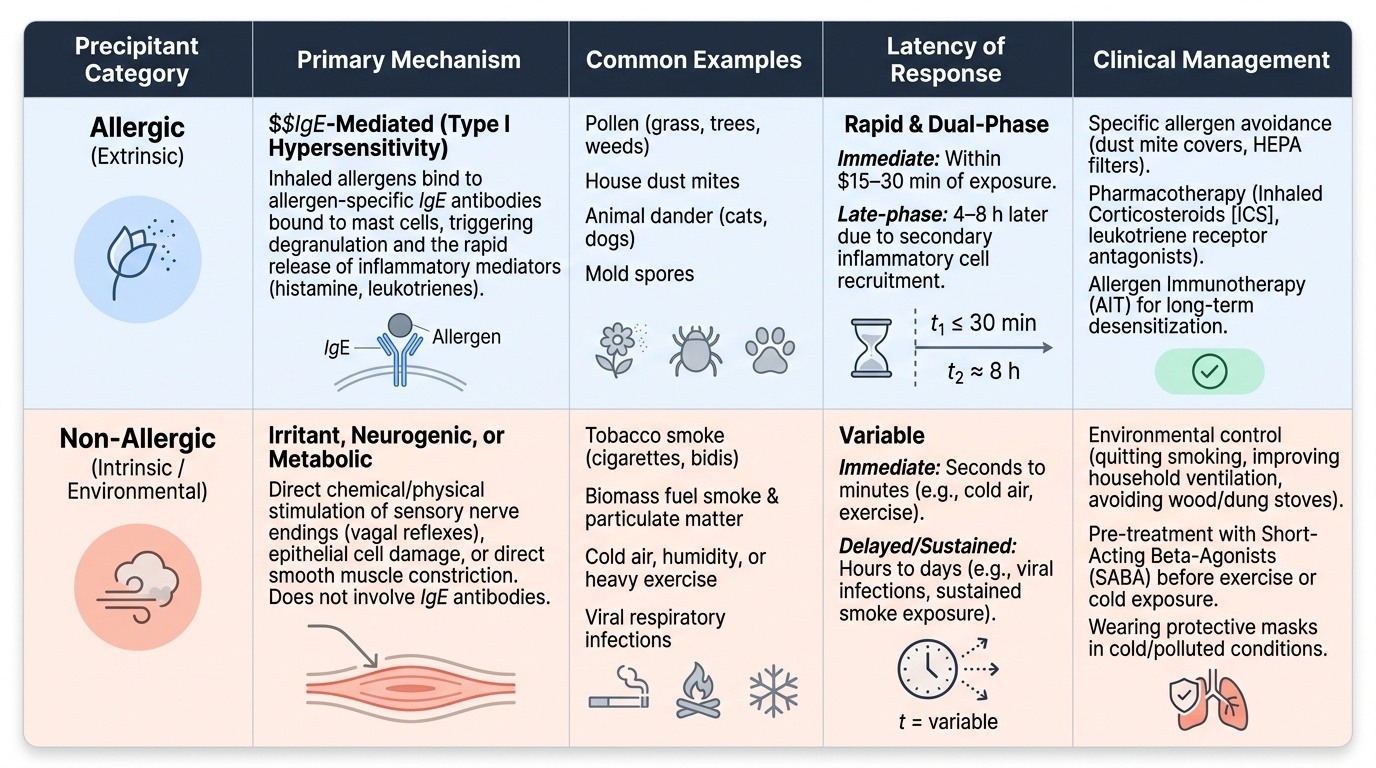
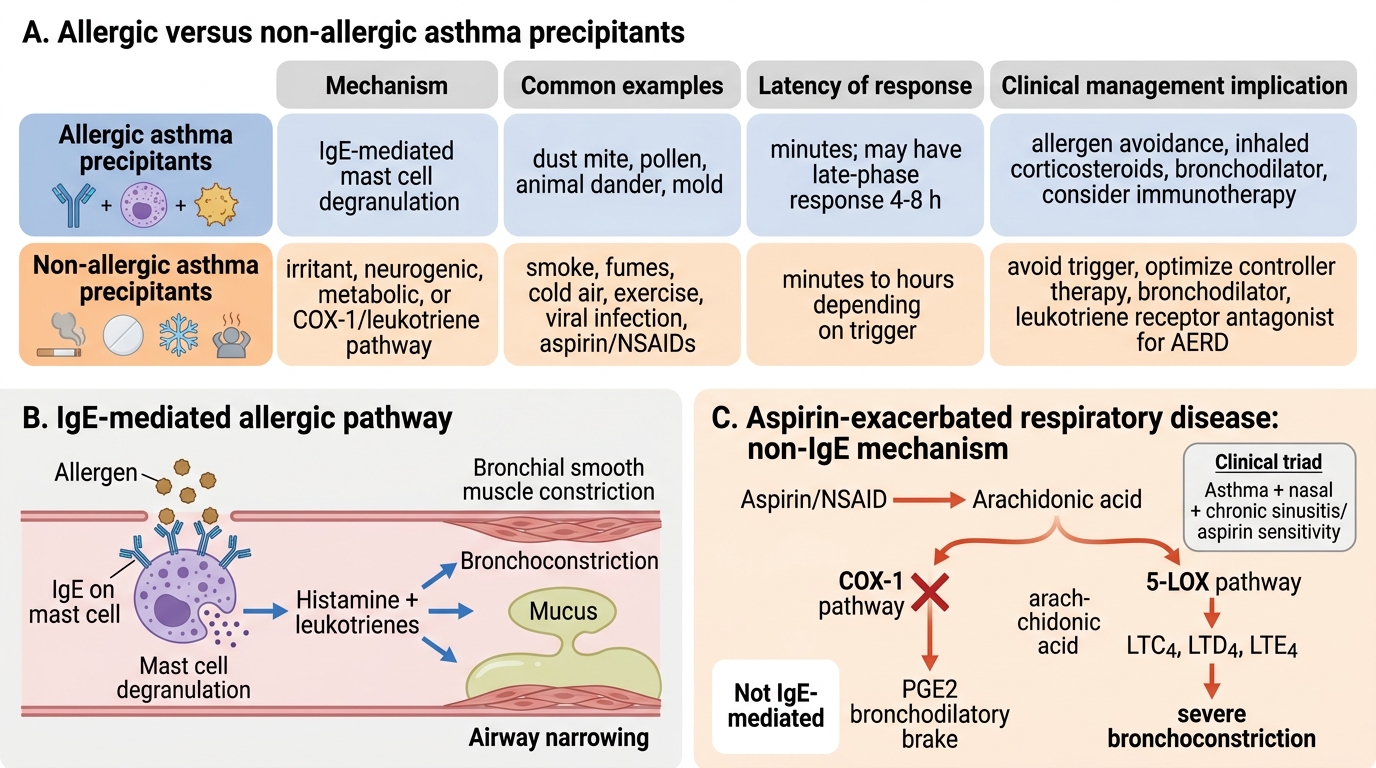
Allergic precipitants of asthma (IgE-mediated, early-phase mechanism):
Sensitised individuals develop specific IgE antibodies directed against aeroallergens. When the allergen binds to IgE on mast cells in the airway mucosa, mast cell degranulation releases preformed mediators (histamine, tryptase, heparin) within minutes, causing immediate bronchospasm, mucosal oedema, and increased mucus secretion — the early-phase asthmatic response. This is followed 4–8 hours later by a late-phase response driven by recruited eosinophils and Th2 lymphocytes releasing IL-4, IL-5, and IL-13, which sustain and amplify the inflammation. Key allergic triggers in the Indian context:
- House dust mite (Dermatophagoides pteronyssinus and D. farinae) — the dominant trigger in urban Indian asthma; thrives in soft furnishing, mattresses, and pillows in humid environments
- Cockroach allergen (Bla g 2) — highly prevalent in urban Indian homes and a major risk factor for paediatric asthma
- Aspergillus mould — triggers both allergic asthma and the more severe allergic bronchopulmonary aspergillosis (ABPA), which requires specific treatment
- Pollen — seasonal variations; grass pollen in post-monsoon; Parthenium hysterophorus (congress grass) is a major contributor in central India
- Animal dander (dog, cat, birds)
Non-allergic precipitants (no IgE mechanism):
- Exercise — exercise-induced bronchoconstriction through airway cooling, drying, and osmolarity changes; treat with pre-exercise SABA or LABA
- Cold air — mechanism similar to exercise; triggers rapid shallow breathing and increased dead-space ventilation
- Aspirin and NSAIDs — AERD (Samter's triad): leukotriene-mediated mechanism via COX-1 inhibition, not IgE; affects approximately 10% of adult asthmatics
- Respiratory infections — particularly viral (rhinovirus); the largest single cause of exacerbations in asthma and COPD
- Emotional stress — neurogenic bronchoconstriction via cholinergic pathways; stress activates the parasympathetic nervous system, releasing acetylcholine and causing smooth muscle contraction
- Occupational sensitisers (acting via non-IgE pathways) — some agents (e.g., isocyanates after the sensitisation phase) cause asthma via irritant or non-IgE mechanisms
- Air pollutants at high levels — ozone, SO₂, NO₂ act directly as irritants on airway epithelium, independent of IgE
- Pregnancy — asthma control changes unpredictably in pregnancy; one-third worsen, one-third improve, one-third remain unchanged; close monitoring is essential
Allergic vs Non-Allergic Asthma Precipitants
SELF-CHECK
A 35-year-old woman develops severe bronchoconstriction within 30 minutes of taking an analgesic for a headache. She has a history of nasal polyps and chronic sinusitis. Skin prick testing for common aeroallergens is negative. What is the most likely mechanism of her asthma exacerbation?
A. IgE-mediated mast cell degranulation triggered by aspirin-specific IgE
B. Inhibition of COX-1 diverting arachidonic acid toward leukotriene synthesis
C. Direct histamine release from basophils by aspirin
D. Aspirin causing excessive prostaglandin E₂ production causing bronchospasm
Reveal Answer
Answer: B. Inhibition of COX-1 diverting arachidonic acid toward leukotriene synthesis
This is aspirin-exacerbated respiratory disease (AERD, Samter's triad: asthma + nasal polyps + aspirin sensitivity). The mechanism is COX-1 inhibition by aspirin and NSAIDs, which blocks the conversion of arachidonic acid to prostaglandins (including the bronchodilatory PGE₂). This diverts arachidonic acid toward the 5-lipoxygenase (5-LOX) pathway, leading to overproduction of cysteinyl leukotrienes (LTC4, LTD4, LTE4), which are potent bronchoconstrictors and pro-inflammatory mediators. This is NOT an IgE-mediated (allergic) mechanism — specific IgE to aspirin does not exist; skin tests are negative. Aspirin does not directly release histamine from basophils in this condition; leukotriene overproduction is the correct mechanism.
CLINICAL PEARL
The fixed-ratio GOLD criterion (FEV1/FVC < 0.70 post-bronchodilator) for COPD diagnosis is the most tested threshold in examinations, but it has a practical limitation: in older adults (>70 years), the FEV1/FVC naturally declines due to age-related loss of elastic recoil, meaning a 0.69 ratio in a 75-year-old may represent normal ageing rather than COPD. The lower limit of normal (LLN) approach, which corrects for age and sex, is more accurate but requires reference equations. For examination purposes and clinical practice in younger patients, remember the fixed 0.70 threshold. The second pearl: the FEV1/FVC ratio for asthma (reversible obstruction) requires documentation of bronchodilator reversibility — an increase in FEV1 of ≥12% AND ≥200 mL simultaneously (not one without the other). A 15% rise on a baseline FEV1 of 1.2 L is only 180 mL — that does not meet both criteria.