Page 2 of 19
IM9.1-2 | Anaemia Foundations — SDL Guide (Part 2)
Iron Deficiency Anaemia — Aetiology and Morphology
Iron deficiency anaemia (IDA) is the commonest nutritional deficiency disorder globally and the leading cause of anaemia in India, accounting for approximately 50% of all anaemia cases in reproductive-age women and children. Iron deficiency develops through three sequential stages: pre-latent (depleted iron stores — low ferritin — but normal serum iron, TIBC, and Hb); latent (iron-deficient erythropoiesis — low ferritin, low serum iron, elevated TIBC, reduced transferrin saturation, but haemoglobin still normal); and overt iron deficiency anaemia (low Hb, low MCV and MCH, high RDW, microcytic hypochromic peripheral film).
The peripheral blood film in IDA shows microcytes (small RBCs), hypochromic cells (increased central pallor, occupying >1/3 of the cell diameter), pencil cells (elliptocytes), and target cells. Poikilocytosis (varied shapes) is common. The characteristic combination of pencil cells and hypochromic microcytes on the film is highly suggestive of IDA.
Aetiology in India:
- Dietary deficiency: staple cereal-based diets low in haem iron (the most bioavailable form), high in phytates (cereals, legumes) and tannins (tea) that inhibit non-haem iron absorption. Vegetarian and vegan diets lack haem iron entirely.
- Increased demand: pregnancy, lactation, rapid growth in infancy and adolescence.
- Chronic blood loss (the most important cause in adults): menorrhagia (the leading cause in premenopausal women), hookworm infestation (Ancylostoma duodenale causes 2–5 mL blood loss per 100 worms per day — a major cause in rural India), peptic ulcer disease, colorectal carcinoma, haemorrhoids, chronic NSAIDs use.
- Malabsorption: coeliac disease, post-gastrectomy, Helicobacter pylori-associated gastric mucosal atrophy.
In any male or postmenopausal female presenting with IDA, GI malignancy must be actively excluded regardless of other explanations — this is a high-yield clinical rule.
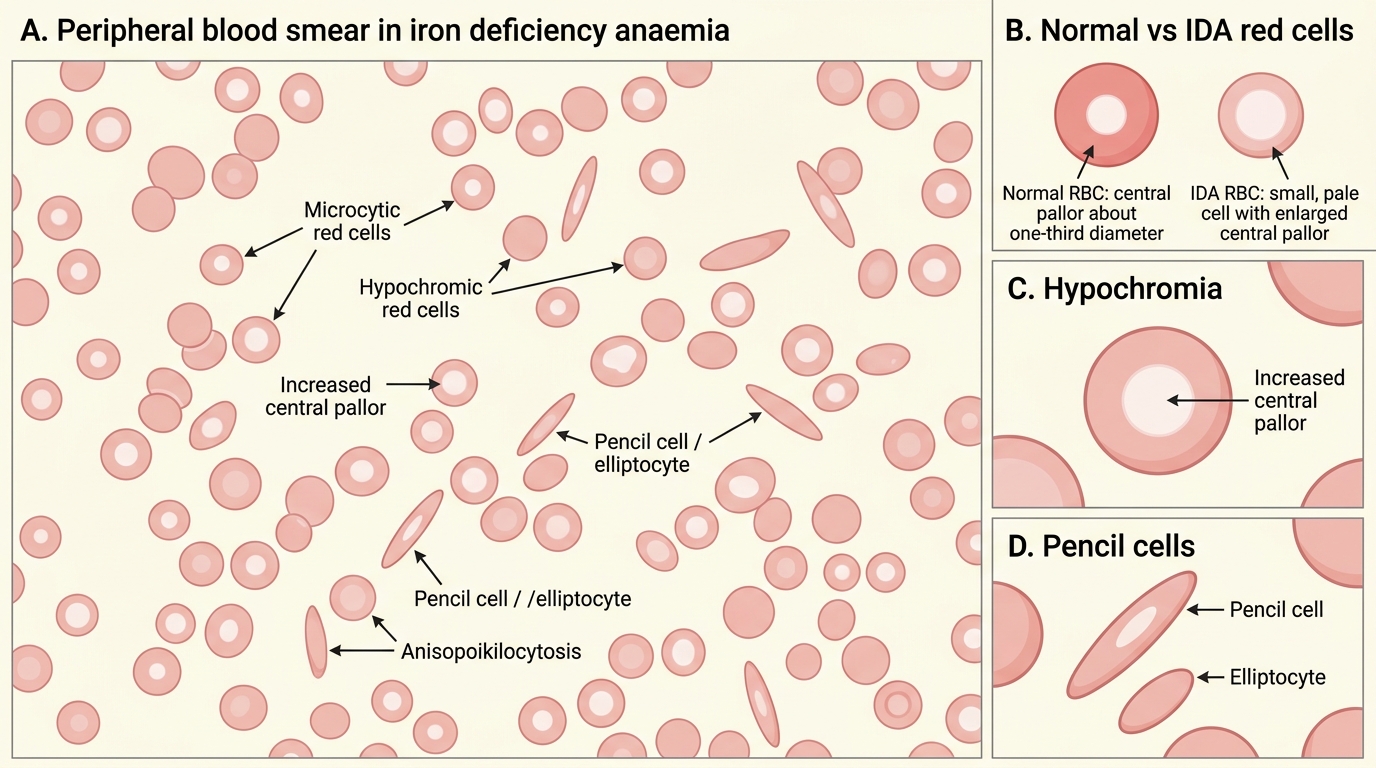
Peripheral Blood Smear in Iron Deficiency Anaemia
Megaloblastic Anaemia — B12 and Folate Deficiency
Megaloblastic anaemia results from impaired DNA synthesis in erythroid precursors due to deficiency of vitamin B12 (cobalamin) or folate, both of which are required as cofactors for the conversion of deoxyuridine monophosphate (dUMP) to deoxythymidine monophosphate (dTMP) — the rate-limiting step in DNA synthesis mediated by thymidylate synthase. When DNA replication is impaired, cells continue to grow (RNA and protein synthesis are unaffected) but fail to divide on schedule, producing large, abnormal red cell precursors called megaloblasts in the bone marrow. The mature red cells released are large, oval erythrocytes — macro-ovalocytes — with MCV typically >100 fL and often >115 fL in established deficiency. The hallmark of megaloblastic change on the peripheral blood film is hypersegmented neutrophils — defined as ≥5% of neutrophils having ≥5 lobes, or any neutrophil having ≥6 lobes.
Vitamin B12 deficiency: Dietary B12 (found exclusively in animal products — meat, fish, eggs, dairy) requires binding to intrinsic factor (IF), secreted by gastric parietal cells, for absorption in the terminal ileum. The primary causes in India are: vegetarian/vegan diet (by far the most prevalent cause — India's predominantly vegetarian population is at high risk, particularly those who consume little dairy or eggs); pernicious anaemia (autoimmune atrophic gastritis causing failure of IF secretion — commonest autoimmune cause; associated with anti-parietal cell antibodies in 90% and anti-IF antibodies in 50–70%); terminal ileal disease (Crohn's disease, ileal resection, tropical sprue — common in South Asia); and gastrectomy. Importantly, B12 has a large hepatic store (2–5 mg) sufficient for 3–4 years, so deficiency develops slowly despite inadequate intake.
The neurological manifestations of B12 deficiency distinguish it sharply from folate deficiency and are clinically critical. Subacute combined degeneration of the spinal cord (SACD) results from demyelination of the dorsal columns (proprioception, vibration sense → sensory ataxia, Romberg positive) and lateral corticospinal tracts (upper motor neurone signs — spasticity, extensor plantar response). Additional neuropsychiatric features: peripheral neuropathy (glove-and-stocking sensory loss), optic neuropathy, dementia, and mood disturbances. SACD can occur in the absence of significant anaemia — B12 deficiency must be considered in any patient with unexplained neuropathy or cognitive decline, especially elderly Indians. Folate deficiency does NOT cause SACD.
Folate deficiency: Dietary folate (found in green leafy vegetables, pulses, fruits) is absorbed in the proximal jejunum. Body stores are limited (5–10 mg — sufficient for only 3–4 months), so deficiency develops faster than B12. Causes: poor dietary intake (the most common — common in poverty, alcoholism, neglect of green vegetables); increased demand (pregnancy and lactation — neural tube defects in the foetus if periconceptional folate supplementation is inadequate, a major Indian public health concern); malabsorption (coeliac disease, tropical sprue); and drugs that interfere with folate metabolism (methotrexate, trimethoprim, phenytoin, phenobarbitone, pyrimethamine). Folate supplementation (5 mg/day) is mandatory in pregnancy; periconceptional folic acid (400–800 µg/day from at least 4 weeks before conception and through 12 weeks of pregnancy) reduces neural tube defect risk by approximately 70%.
Biochemical distinction between B12 and folate deficiency: serum B12 <200 pg/mL (normal 200–900 pg/mL) and serum folate <3 ng/mL (normal >5 ng/mL) are primary markers. However, both can be borderline. Serum methylmalonic acid (MMA) is elevated specifically in B12 deficiency (B12 is a cofactor for MMA metabolism) — normal folate does NOT affect MMA. Homocysteine is elevated in BOTH B12 and folate deficiency (both are required for homocysteine remethylation to methionine). MMA therefore discriminates B12 from folate deficiency and is the confirmatory test when serum B12 is borderline.
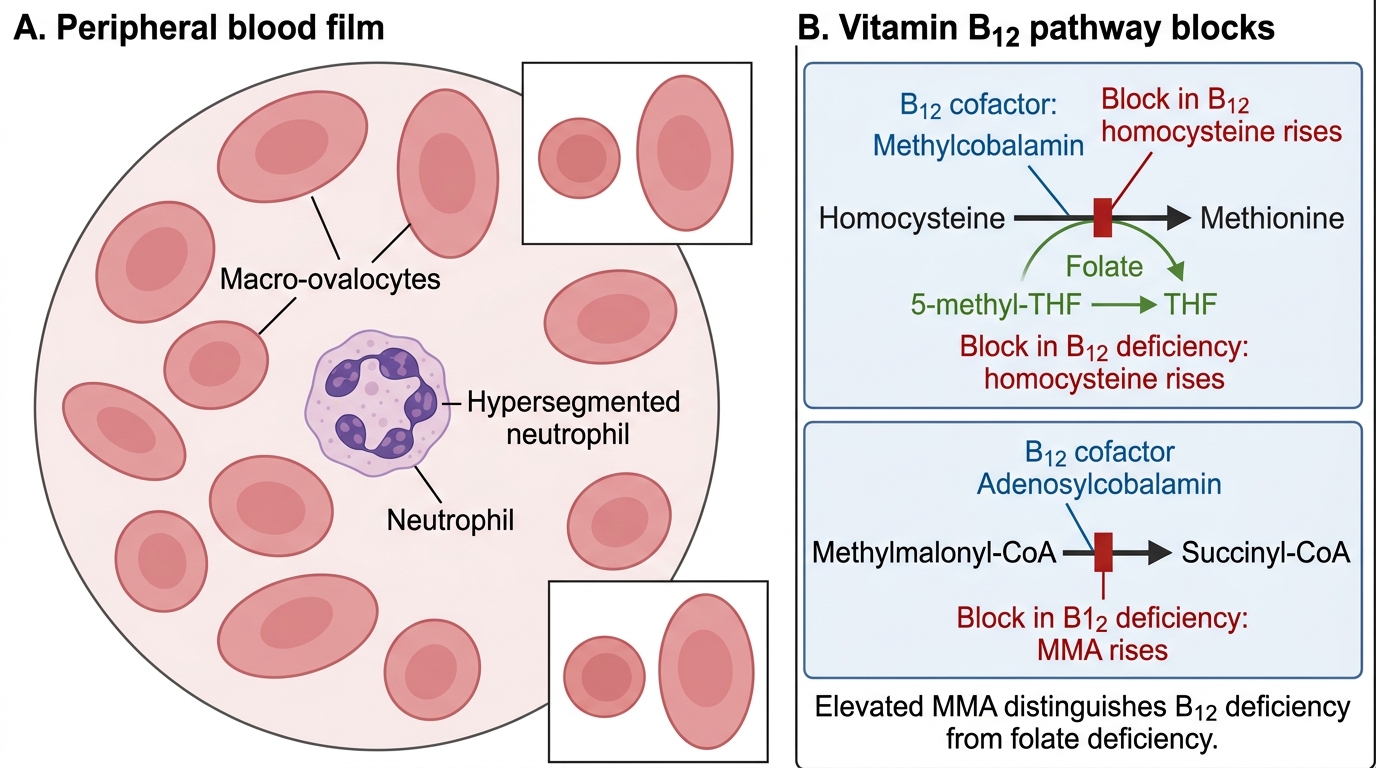
Megaloblastic Anaemia and Vitamin B12 Pathway Blocks
SELF-CHECK
A 60-year-old vegetarian man presents with 6 months of difficulty walking and memory decline. His Hb is 11.2 g/dL, MCV 112 fL. Peripheral film shows macro-ovalocytes and hypersegmented neutrophils. Serum B12 is 158 pg/mL. Which single biochemical test best CONFIRMS B12 deficiency rather than folate deficiency as the cause?
A. Serum folate level
B. Serum homocysteine level
C. Serum methylmalonic acid (MMA) level
D. Anti-intrinsic factor antibody
Reveal Answer
Answer: C. Serum methylmalonic acid (MMA) level
Serum methylmalonic acid (MMA) is elevated specifically in vitamin B12 deficiency because B12 (adenosylcobalamin) is a required cofactor for the conversion of methylmalonyl-CoA to succinyl-CoA. Folate deficiency does NOT elevate MMA. Homocysteine is elevated in BOTH B12 and folate deficiency (both are cofactors for homocysteine remethylation to methionine) — so it does not discriminate between the two. Anti-intrinsic factor antibody is specific for pernicious anaemia (the autoimmune cause of B12 deficiency) but does not confirm B12 deficiency in a vegetarian. Elevated MMA confirms B12 deficiency as the metabolic cause.
Haemolytic Anaemias — Mechanisms and Classification
Haemolytic anaemia is defined by premature destruction of red blood cells before their normal 120-day lifespan, causing accelerated RBC turnover. The marrow's compensatory reticulocytosis (typically 5–20%) produces the elevated reticulocyte index that distinguishes haemolytic from hypoproliferative causes. All haemolytic anaemias share a common biochemical fingerprint of accelerated RBC destruction: elevated serum lactate dehydrogenase (LDH) — released from lysed erythrocytes; reduced or absent serum haptoglobin — a plasma protein that binds free haemoglobin released by intravascular haemolysis (haptoglobin is consumed when it binds free Hb and the complex is cleared by the liver); elevated unconjugated (indirect) bilirubin — from haem catabolism via the reticuloendothelial system; and elevated reticulocyte count.
The primary classification of haemolytic anaemia is by site of destruction: intravascular (RBC lysis within blood vessels — releases free Hb into plasma, causing haemoglobinaemia, haemoglobinuria — 'Coca-Cola urine', and markedly reduced haptoglobin; causes: mismatched blood transfusion, PNH, G6PD acute haemolytic crisis, microangiopathic haemolytic anaemia) versus extravascular (macrophage-mediated phagocytosis in spleen and liver — the predominant mechanism in most haemolytic anaemias; free Hb not released into plasma; haptoglobin reduction is milder; splenomegaly common). The second classification axis is intrinsic (RBC defect itself) versus extrinsic (normal RBCs destroyed by abnormal external forces).
Intrinsic haemolytic anaemias — India-relevant:
- Sickle cell disease (SCD): autosomal recessive mutation (β-globin gene, chromosome 11 — glutamic acid → valine at position 6, HbS). Under low oxygen tension, HbS polymerises into rigid fibres that distort the RBC into the characteristic sickle shape. HbSS (homozygous) — severe haemolytic anaemia, vaso-occlusive crises (pain crises, acute chest syndrome, stroke), splenic autoinfarction → functional asplenia → susceptibility to encapsulated bacteria. Highly prevalent in central India (Chhattisgarh, Maharashtra tribal belt — carrier frequency up to 35% in some communities), Odisha, Gujarat. Diagnosed by haemoglobin electrophoresis (HPLC) — HbS band.
- Thalassaemias: autosomal recessive reduction or absence of α-globin (α-thalassaemia) or β-globin (β-thalassaemia) chain synthesis. β-thalassaemia major (Cooley's anaemia) presents in infancy with severe transfusion-dependent haemolytic anaemia, extramedullary haematopoiesis (bossing of skull, chipmunk facies, hepatosplenomegaly), iron overload from transfusions. β-thalassaemia trait (minor) is asymptomatic with mild microcytosis (MCV 60–75 fL), normal RDW, normal or mildly reduced Hb — distinguished from IDA by NORMAL serum ferritin and HIGH HbA2 (>3.5%) on HPLC. India is the thalassaemia capital of the world — approximately 40 million carriers, 10,000–15,000 new thalassaemia major births per year.
- G6PD deficiency: X-linked recessive deficiency of glucose-6-phosphate dehydrogenase, an enzyme of the hexose monophosphate shunt critical for generating NADPH — the primary antioxidant defence of the RBC (which lacks mitochondria and depends entirely on NADPH to reduce glutathione). In the absence of G6PD activity, oxidative stress (from drugs — primaquine, dapsone, nitrofurantoin; fava beans; infections) causes Heinz body formation (denatured haemoglobin aggregates) and acute haemolytic episodes. In India, G6PD deficiency is prevalent (7–8% of males), particularly the Mediterranean and Viangchan variants. Neonatal jaundice can be severe. Haemolytic episodes are self-limited; management is avoidance of triggers.
- Hereditary spherocytosis: autosomal dominant defect in RBC membrane proteins (spectrin, ankyrin, band 3) → loss of membrane surface area → spherical cells → increased splenic sequestration and destruction. Peripheral film shows spherocytes (small, dark, no central pallor) + reticulocytosis. Osmotic fragility test (fragility increases) and eosin-5-maleimide (EMA) binding test (reduced binding) confirm the diagnosis. Splenectomy is the definitive treatment in severe cases.
Extrinsic haemolytic anaemias:
- Autoimmune haemolytic anaemia (AIHA): autoantibodies directed against RBC surface antigens. Classified by thermal reactivity: warm AIHA (IgG antibodies active at 37°C — the most common type; associated with SLE, CLL, drugs, idiopathic; detected by direct Coombs/direct antiglobulin test — DAT which detects IgG or complement coated on RBC surface); cold AIHA (IgM antibodies active below 32°C — cold agglutinin disease, associated with Mycoplasma pneumoniae, EBV, lymphomas). DAT positive in all true AIHA.
- Microangiopathic haemolytic anaemia (MAHA): mechanical fragmentation of RBCs in narrowed/thrombosed small vessels → schistocytes and helmet cells on film. Causes include TTP (thrombotic thrombocytopenic purpura — ADAMTS13 deficiency), HUS (haemolytic uraemic syndrome — Shiga-toxin E. coli), DIC, malignant hypertension, HELLP syndrome.
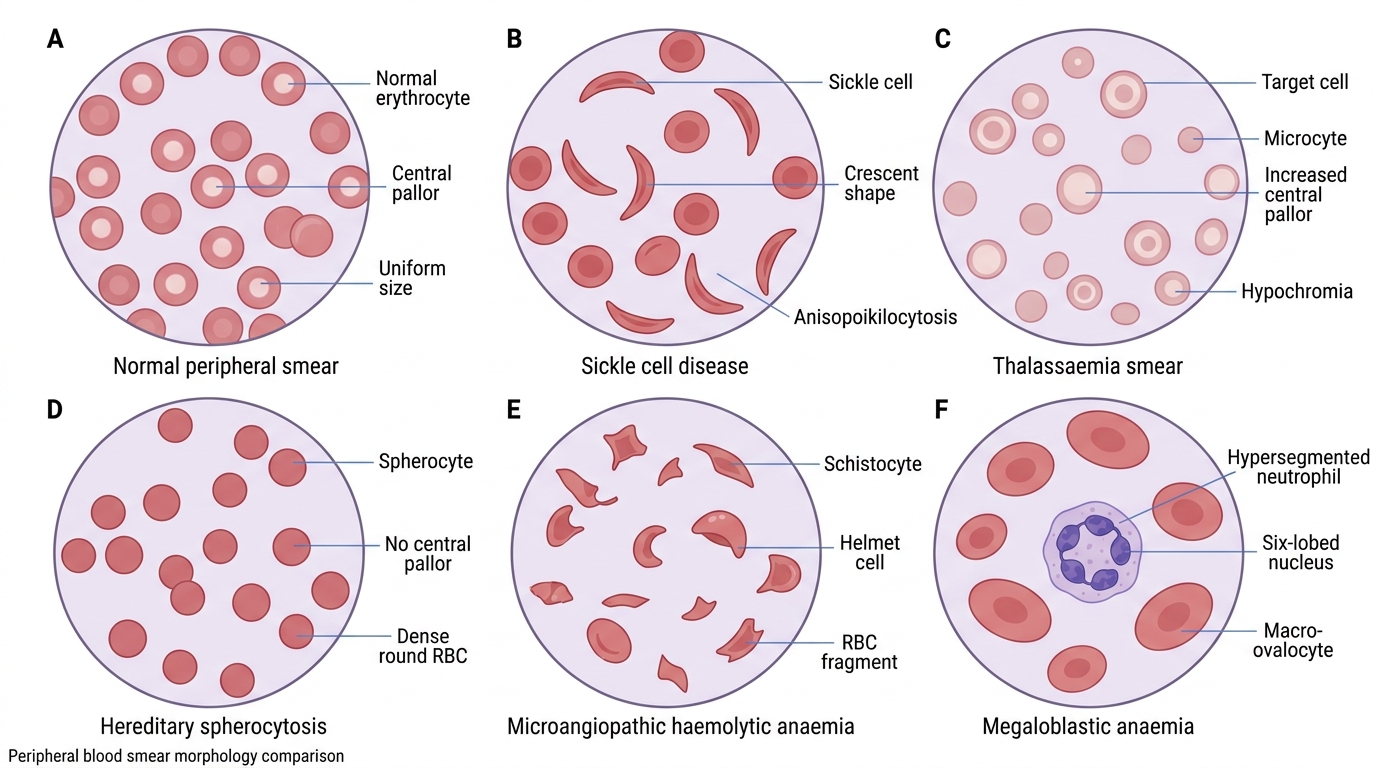
Peripheral Blood Smear Patterns in Anaemia