Page 4 of 33
OP4.2 | Congenital Corneal Anomalies and Corneal Inflammations — SDL Guide
Learning Objectives
- Enumerate the major congenital corneal anomalies and describe their clinical features and associations
- Differentiate megalocornea from buphthalmos and keratoconus from other ectasias
- Classify corneal inflammations by depth and aetiology
- Describe the clinical features and causes of interstitial keratitis including Hutchinson's triad
- Outline the investigation and management principles for congenital corneal anomalies and corneal inflammations
INSTRUCTIONS
Congenital corneal anomalies range from simple size variations to complex structural failures involving multiple layers. Corneal inflammations (keratitis) span superficial infections to deep autoimmune stromal disease. Both categories demand a systematic approach: identify the pattern, correlate with associations or risk factors, and match to the appropriate investigation and treatment pathway. This module builds that framework for the MBBS final-year student entering clinical postings.
References
- Khurana AK. Comprehensive Ophthalmology, 7th ed. Ch 5: Diseases of the Cornea (textbook)
- Parsons' Diseases of the Eye, 23rd ed. Ch 9: The Cornea (textbook)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A 2-year-old boy is brought by his mother because his eyes 'look bigger than other children's'. His horizontal corneal diameter measures 14.5 mm bilaterally. She says he sees well and is not bothered by light. You check his intraocular pressure: 14 mmHg in both eyes. The anterior chambers are deep and symmetric. You examine the corneas with a torch and find them clear, without any horizontal striations. Meanwhile, in the adjacent clinic room, a 3-year-old with similar-looking large corneas is crying and photophobic, with cloudy corneas and a pressure of 30 mmHg. Same appearance at first glance; completely different diagnosis, urgency, and outcome.
WHY THIS MATTERS
Recognising congenital corneal anomalies at their first presentation determines whether a child undergoes urgent intervention (congenital glaucoma, Peters' anomaly threatening vision) or appropriate surveillance (megalocornea). Missing these diagnoses costs patients their vision — amblyopia is irreversible if not treated in the critical period. Equally, interstitial keratitis is a rare but high-yield presentation because it signals a treatable systemic disease (syphilis, TB) and the ophthalmological findings may be the first clue. These are classic examination questions precisely because they test pattern recognition across systems.
RECALL
From embryology: the corneal epithelium derives from surface ectoderm; stroma and endothelium from neural crest cells. The corneal stroma develops in the 5th–8th week of gestation; any failure of neural crest migration produces structural anomalies. From the previous module (SDL 1), recall that Descemet's membrane is remarkably resistant to collagenases — its absence is a defining feature of Peters' anomaly. Recall also that Haab's striae are horizontal breaks in Descemet's membrane caused by stretching in buphthalmos — the directionality helps you distinguish them from the vertical/oblique Vogt's striae of keratoconus.
Overview and Clinical Presentation of Congenital Corneal Anomalies
Congenital corneal anomalies present in infancy or early childhood with a limited repertoire of symptoms: reduced vision (the infant does not fix and follow), abnormal corneal size (too small, too large, or abnormal shape), opacity, or photophobia. The key to early diagnosis is examining any infant whose parents report unusual eye appearance, and any newborn with abnormal eye size, cloudiness, or photophobia — these are not benign findings.
The conditions can be grouped into three categories for systematic learning: anomalies of size and curvature (microcornea, megalocornea, keratoconus, keratoglobus), embryological structural defects (Peters' anomaly, sclerocornea), and isolated transient conditions such as birth-related trauma. The distinction between these categories influences the prognosis and treatment approach fundamentally. A size anomaly with clear corneas may need nothing more than refractive correction; a structural defect involving Descemet's membrane and the endothelium may need keratoplasty in infancy to prevent dense amblyopia.
The first and most important diagnostic step in any child with a large cornea is measuring the intraocular pressure. Buphthalmos (ox eye) is the enlarged, stretched cornea of congenital glaucoma — a sight-threatening emergency requiring urgent surgery. Before labelling any large cornea as a benign variant (megalocornea), you must exclude elevated IOP, Haab's striae, and corneal oedema.
Size and Curvature Anomalies: Microcornea, Megalocornea, and Keratoconus
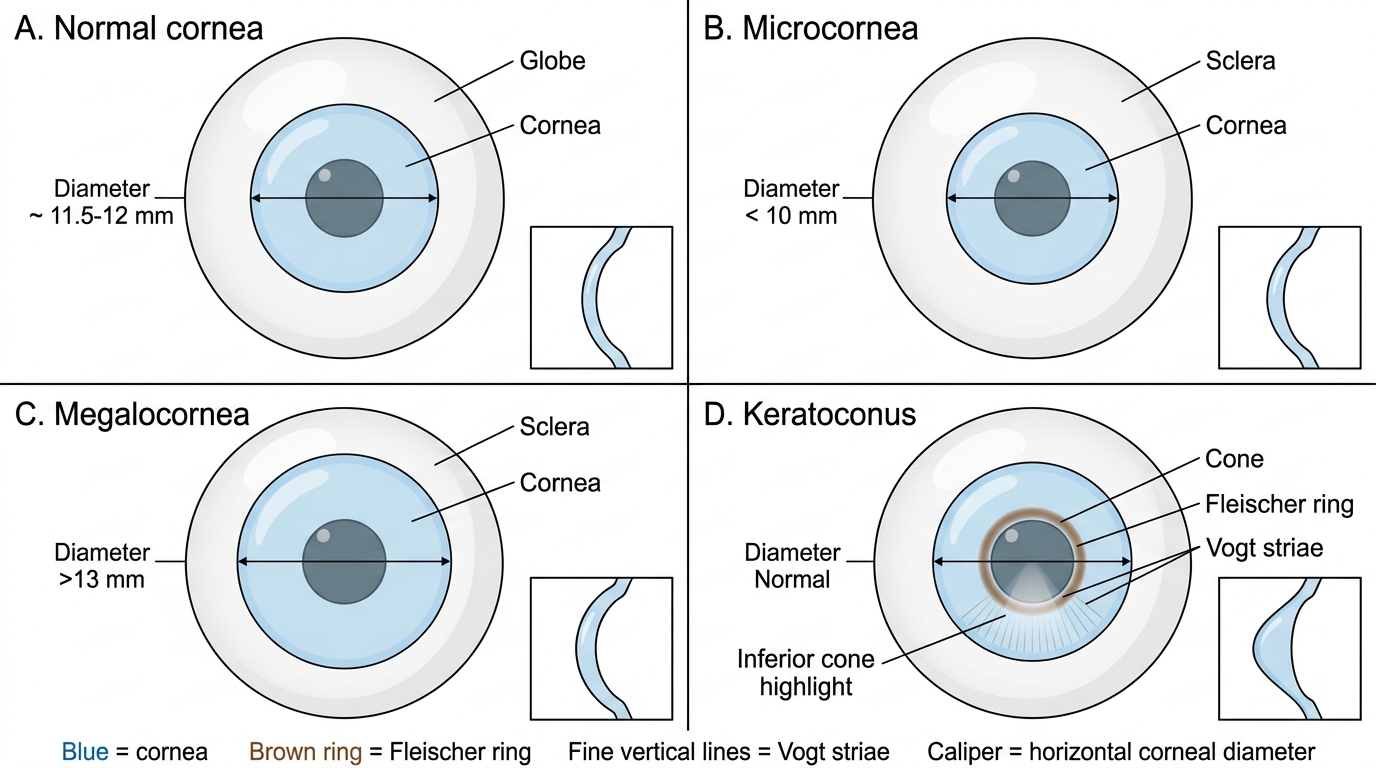
Microcornea is defined as a horizontal corneal diameter less than 10 mm (normal adult: 11.5–12 mm). The cornea may be structurally normal or there may be associated anomalies: microphthalmia (the whole eye is small), congenital cataract, anterior chamber angle anomalies predisposing to glaucoma, and high hypermetropia. The condition may be unilateral or bilateral. Management includes refractive correction, amblyopia treatment, and monitoring for glaucoma.
Megalocornea is a horizontal diameter exceeding 13 mm with a clear cornea and normal IOP. It is most often a benign X-linked recessive condition (gene on Xq21.3–q22), predominantly in males. The corneas are optically clear, deepen the anterior chamber, and the patient may have a degree of arcus juvenilis or lens subluxation (ectopia lentis) in adulthood. The critical clinical task is distinguishing megalocornea from buphthalmos: in buphthalmos (congenital glaucoma), the IOP is elevated, the cornea is oedematous/cloudy, and Haab's striae — horizontal breaks in Descemet's membrane from stretching — are pathognomonic. In megalocornea, IOP is normal and no Haab's striae are present.
Keratoconus is a non-inflammatory, bilateral, progressive thinning and conical ectasia of the cornea, most often beginning at puberty and progressing through the 3rd–4th decade when it typically stabilises. It is the commonest corneal ectasia worldwide. The cone develops in the paracentral region, distorting the regular collagen lattice described in SDL 1. This produces progressive irregular astigmatism, reducing corrected visual acuity even with spectacles. Three classic signs: (i) Fleischer's ring — a brownish ring of iron (haemosiderin) deposition in the epithelial basal cells at the base of the cone, best seen with a cobalt blue filter; (ii) Vogt's striae — fine vertical or oblique stress lines in the deep stroma, which disappear on gentle pressure to the eye; (iii) Munson's sign — an outward V-shaped protrusion of the lower eyelid in downgaze, reflecting the conical shape. Advanced keratoconus may develop acute hydrops — a sudden rupture of Descemet's membrane allowing aqueous humour to flood the stroma, causing dramatic corneal oedema and pain. Management: early stages with spectacles/rigid contact lenses; halted by corneal collagen cross-linking (CXL) with riboflavin + UV-A (stiffens collagen lamellae); advanced disease requiring keratoplasty (DALK or PK).
Keratoglobus is a rarer spherical ectasia involving the entire cornea (unlike the central/paracentral cone of keratoconus), with thinning maximal at the periphery. It is associated with connective tissue disorders (Ehlers-Danlos syndrome, Marfan syndrome) and Leber's congenital amaurosis.
Comparison of Corneal Shape Abnormalities
SELF-CHECK
A 14-year-old boy presents with progressive blurring in both eyes not correctable with spectacles. Slit-lamp shows Fleischer's ring and Vogt's striae in both corneas. Corneal topography shows paracentral inferior steepening. What is the most appropriate next management step after diagnosis?
A. Urgent penetrating keratoplasty (PK) to prevent further ectasia
B. Corneal collagen cross-linking (CXL) with riboflavin and UV-A to halt progression
C. Topical corticosteroids to reduce inflammation
D. Refer for LASIK refractive correction to address the irregular astigmatism
Reveal Answer
Answer: B. Corneal collagen cross-linking (CXL) with riboflavin and UV-A to halt progression
Corneal collagen cross-linking (CXL) is the standard of care for progressive keratoconus. It stiffens the collagen lamellae by forming covalent bonds between collagen fibrils, halting progression. LASIK is absolutely contraindicated in keratoconus as it further weakens the cornea and can precipitate ectasia. Corticosteroids have no role. Keratoplasty is reserved for advanced cases where contact lens correction has failed.
Structural Embryological Anomalies: Peters Anomaly and Sclerocornea
Peters' anomaly is a central corneal opacity resulting from a defect in embryological development of the posterior corneal layers. The defining pathological finding is a posterior corneal defect: absence of the central Descemet's membrane and endothelium, with or without iridocorneal adhesions (iridocorneal synechia), and in more severe cases, attachment of the crystalline lens to the posterior corneal surface (lenticular-corneal adhesion). The central corneal opacity ('leucocoria of the cornea') is present at birth. Associations include chromosomal abnormalities (deletions at 4p, trisomies), PAX6 gene mutations, PITX2 mutations (Axenfeld-Rieger spectrum), and systemic abnormalities. Treatment requires urgent optical rehabilitation: in mild unilateral cases with partial opacity, amblyopia therapy; in bilateral or dense cases, keratoplasty in infancy (technically challenging, with significant risk of failure, graft rejection, and glaucoma). Prognosis is guarded.
Sclerocornea is a congenital non-progressive opacity in which the peripheral (and sometimes central) cornea is replaced by tissue resembling sclera — white, vascularised, and non-transparent. The limbus is ill-defined. It results from failure of mesenchymal differentiation of the anterior segment. It may be unilateral or bilateral, and may be complete (entire cornea involved) or peripheral only. It can be associated with systemic connective tissue disorders and chromosomal anomalies. Management in unilateral or peripheral cases may be conservative; bilateral complete sclerocornea requires keratoplasty for visual rehabilitation, though outcomes are poor.
The anterior segment dysgenesis spectrum — which includes Peters' anomaly, sclerocornea, Axenfeld's anomaly (posterior embryotoxon with iris strands), and Rieger's anomaly — all result from abnormal neural crest cell migration and differentiation during embryogenesis. All carry a significant risk of secondary glaucoma, which must be monitored lifelong.