Page 31 of 53
PE19.11 | Neonatal Hypocalcemia — SDL Guide
Learning Objectives
- Explain neonatal calcium homeostasis and the physiological transition from intrauterine to extrauterine life
- Distinguish early-onset from late-onset neonatal hypocalcemia by timing, aetiology, and PTH status
- Recognise the clinical and ECG features of neonatal hypocalcemia
- Describe the correct management: IV calcium gluconate with cardiac monitoring for symptomatic cases, oral supplements for mild cases, and correction of underlying hypomagnesaemia
- Identify at-risk neonates and outline preventive strategies
INSTRUCTIONS
Neonatal hypocalcemia is the second most common metabolic emergency after hypoglycemia in the newborn period. Its presentation is easily confused with other neonatal conditions — jitteriness, apnoea, and seizures all overlap with hypoglycemia and sepsis. Getting the aetiology right matters enormously: an infant on cow's milk formula with late-onset hypocalcemia needs a completely different approach from a preterm IDM with early-onset disease. The treatment — intravenous calcium gluconate — is safe only when given slowly with continuous cardiac monitoring; a rapid bolus causes life-threatening bradycardia and cardiac arrest.
References
- Ghai Essential Pediatrics, 9th ed., Ch. 7 — Neonatal Metabolic Disorders (textbook)
- Nelson Textbook of Pediatrics, 21st ed., Ch. 128 — Hypocalcemia (textbook)
- IAP / NNF India — Guidelines on Neonatal Hypocalcemia Management (guideline)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A 5-day-old male infant born at term is brought to the outpatient clinic with a 1-day history of twitching of the face and limbs. The mother mentions she has been supplementing breastfeeding with undiluted cow's milk formula since day 3. On examination, the baby is alert, and gentle tapping over the cheek in front of the ear elicits a brief facial twitch. Serum calcium is reported as 6.8 mg/dL. What is the diagnosis, why did this happen on cow's milk formula, and what must you check and monitor before starting treatment?
WHY THIS MATTERS
Neonatal hypocalcemia occurs in 1–5% of all neonates and in significantly higher proportions of at-risk groups — up to 30–50% of very preterm neonates and infants of diabetic mothers in the early postnatal period. Calcium is the most abundant mineral in the body and is essential for neuromuscular transmission, cardiac contractility, enzyme activation, and skeletal mineralisation. In neonates, even moderate reductions in ionised calcium cause neuromuscular hyperexcitability — jitteriness, seizures, and laryngospasm — that can be life-threatening if misdiagnosed or treated incorrectly. Understanding the two distinct clinical syndromes (early-onset vs late-onset hypocalcemia) is essential because they differ in aetiology, PTH status, phosphate levels, and treatment. Crucially, a rapid intravenous calcium bolus is a potentially fatal intervention; cardiac monitoring is non-negotiable.
RECALL
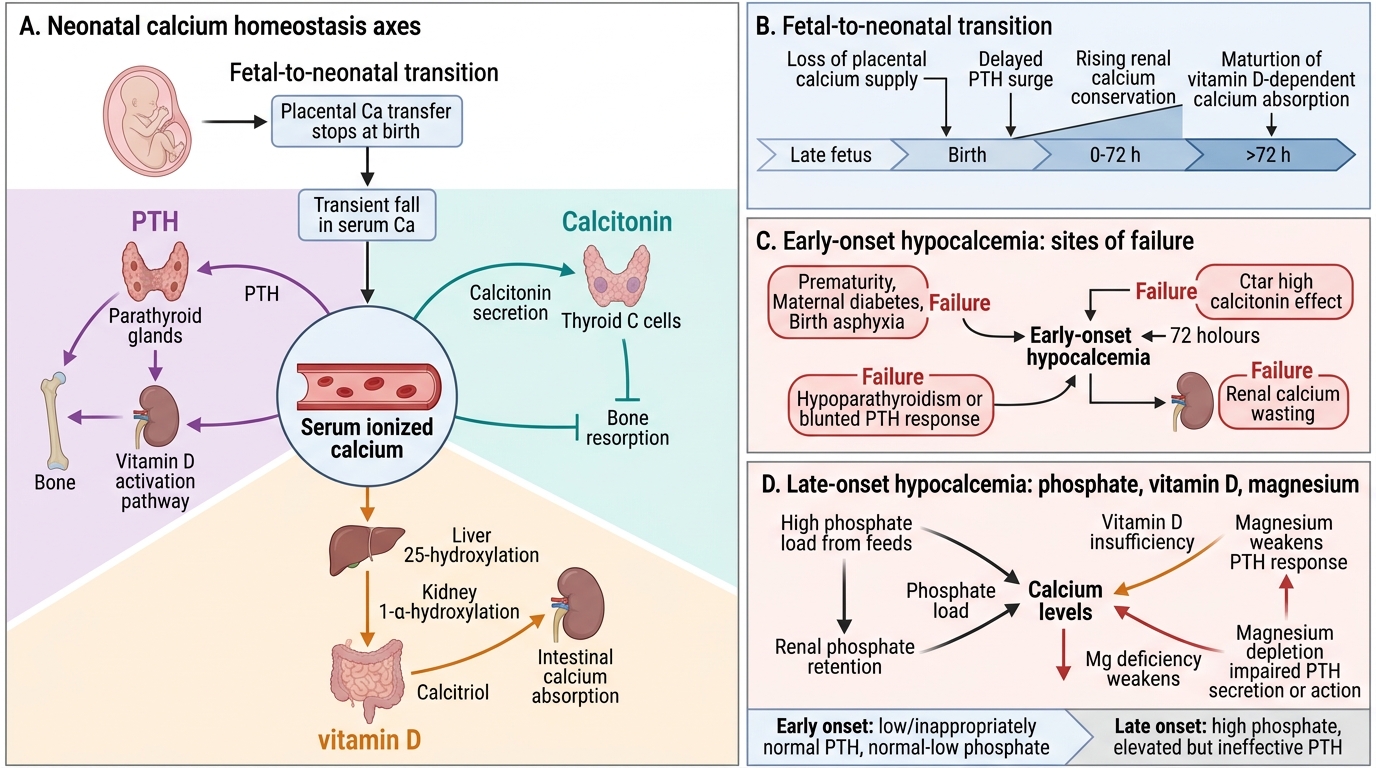
From Physiology, recall that serum calcium exists in three fractions: ionised (free, ~50%; physiologically active), protein-bound (~40%, mainly albumin), and complexed (~10%, citrate/phosphate). Only ionised calcium regulates neuromuscular and cardiac function. Parathyroid hormone (PTH) raises serum calcium via three mechanisms: bone resorption (osteoclast activation), renal calcium reabsorption (distal tubule), and stimulation of renal 1α-hydroxylase to convert 25-hydroxyvitamin D to active 1,25-dihydroxyvitamin D (calcitriol), which increases intestinal calcium absorption. Calcitonin (from thyroid C-cells) opposes PTH, lowering serum calcium by inhibiting osteoclasts. In utero, the fetus receives calcium via active placental transport; fetal serum calcium is maintained above maternal levels (~10.5 vs 9.5 mg/dL). This high fetal calcium suppresses fetal PTH but elevates fetal calcitonin — setting up the neonatal transition as a period of parathyroid gland 'unpriming' that must be rapidly reversed after birth.
Calcium Homeostasis in the Neonate: Physiology and Transition
The fetal-to-neonatal calcium transition is a critical physiological event. In utero, the placenta actively transports calcium from mother to fetus against a concentration gradient, maintaining fetal serum calcium at approximately 10.5–11.0 mg/dL — higher than maternal levels. This sustained fetal hypercalcaemia has two consequences: fetal parathyroid gland activity is chronically suppressed (low PTH), and fetal calcitonin secretion is elevated. At birth, when the placental calcium supply is abruptly cut off, the neonate must rapidly activate the PTH–vitamin D axis to defend serum calcium from falling.
In healthy term neonates, serum calcium falls transiently to approximately 8.0–8.5 mg/dL at 24–48 hours of age (the "physiological nadir"), then rises as PTH secretion is upregulated and oral feeding begins delivering dietary calcium. The ionised calcium fraction is the clinically important measurement — it is unaffected by albumin levels (which change with hydration and nutrition) and directly reflects neuromuscular calcium availability. The normal ionised calcium in a neonate is ≥1.1–1.2 mmol/L; values below 1.0 mmol/L define hypocalcemia irrespective of total calcium.
Several neonatal-specific factors make this transition vulnerable. Preterm neonates have immature parathyroid glands and have not completed the third-trimester phase of peak calcium accretion (85% of fetal calcium is deposited in the last trimester). Infants of diabetic mothers have chronic fetal hyperglycaemia stimulating hypertrophy of all fetal glands, but the resulting IDM parathyroid is functionally immature. Additionally, neonatal calcitonin levels remain elevated for several days post-birth, continuing to suppress bone resorption and reducing the PTH-mediated calcium mobilisation needed to compensate for the loss of placental supply.
⚑ AI image — pending faculty review (auto-QA score 7/10; best of 3 attempts)
Neonatal Calcium Homeostasis and Hypocalcemia
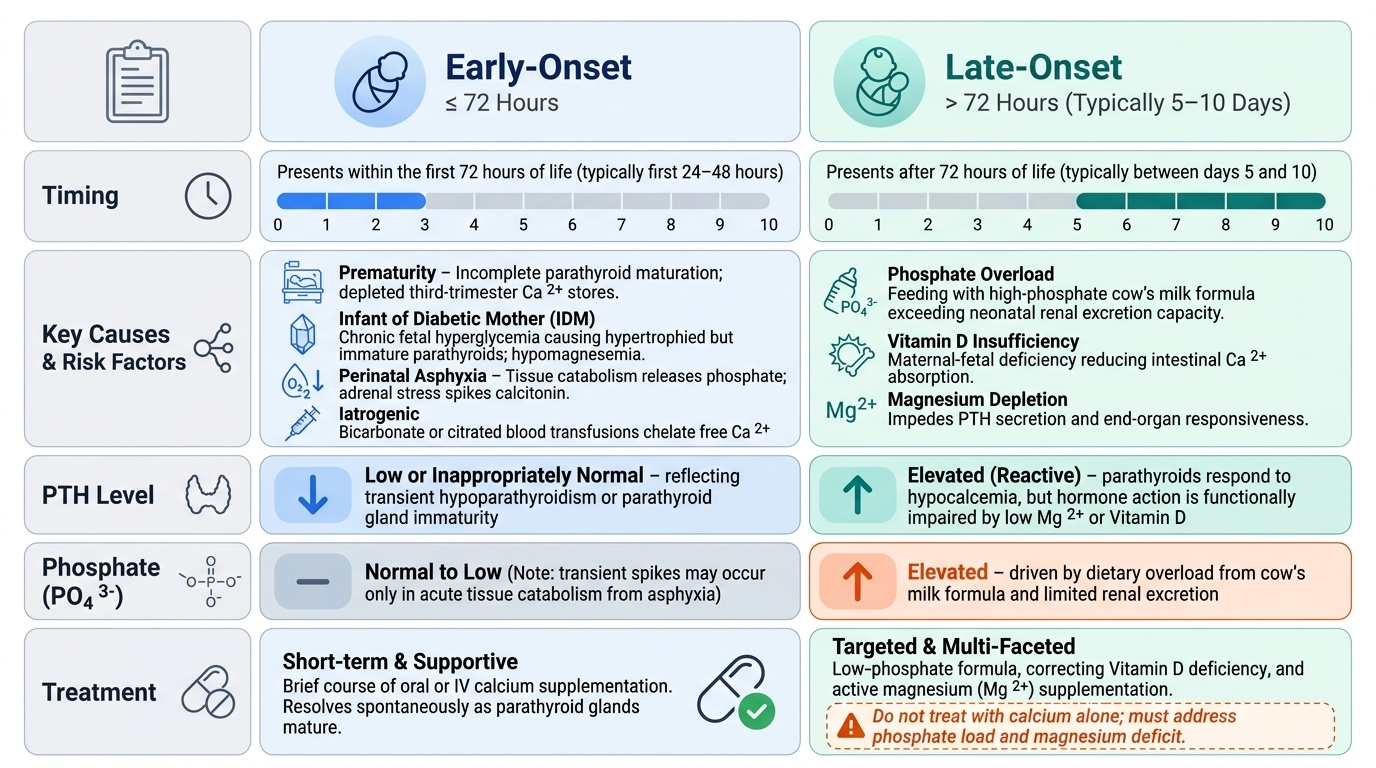
Aetiology: Early-Onset vs Late-Onset Hypocalcemia
Neonatal hypocalcemia is clinically and pathophysiologically divided into two syndromes by the timing of onset. This classification is not merely academic — it directs the aetiological workup and determines whether a simple oral supplementation course will suffice or whether lifelong endocrine therapy is needed. The key distinguishing features are not just timing but also the serum phosphate level and the PTH response: early-onset disease is characterised by low or inappropriately normal PTH with normal-to-low phosphate, while late-onset disease typically features elevated phosphate (from dietary overload or renal retention) and a PTH that is elevated in response but functionally impaired by magnesium depletion or vitamin D insufficiency. Understanding these mechanistic differences prevents the common error of treating late-onset hypocalcemia with calcium supplementation alone while missing the phosphate load or a correctable magnesium deficit.
Provided image
Early-onset hypocalcemia presents within the first 72 hours of life. It arises primarily from transient suppression or functional immaturity of the parathyroid gland, compounded by elevated calcitonin. The key risk groups and mechanisms are:
- Prematurity — incomplete PTH maturation; depleted calcium stores (third-trimester accretion incomplete)
- Infant of diabetic mother (IDM) — chronic fetal hyperglycaemia → hypertrophied but functionally immature parathyroids; hypomagnesaemia (often co-exists, as PTH secretion requires magnesium as a cofactor)
- Perinatal asphyxia — tissue catabolism releases phosphate (chelates calcium); adrenal stress elevates calcitonin; impaired PTH response
- Iatrogenic — excessive bicarbonate use or exchange transfusion with citrated blood chelates ionised calcium acutely
Late-onset hypocalcemia presents after 72 hours, typically at 5–10 days of age. The hallmark mechanism is phosphate overload combined with vitamin D insufficiency:
- High-phosphate cow's milk feeds — cow's milk contains ~6× more phosphate than human breast milk. In a neonate with an immature kidney that cannot efficiently excrete phosphate, the phosphate load raises serum phosphate, which complexes with calcium in the serum, driving ionised calcium down. This is the classic cause in formula-fed or cow's milk-supplemented infants
- Vitamin D deficiency — common in exclusively breastfed infants of vitamin D-deficient mothers; impairs intestinal calcium absorption
- Hypomagnesaemia — magnesium is required for PTH secretion and for PTH receptor signalling at target organs; hypomagnesaemia (<0.6 mmol/L) causes PTH-resistant hypocalcemia that will not respond to calcium therapy alone
- DiGeorge syndrome (22q11.2 deletion) — aplasia or hypoplasia of parathyroid glands → permanent hypoparathyroidism; associated with conotruncal cardiac defects, T-cell immunodeficiency (thymic aplasia), and characteristic facies
- Maternal hyperparathyroidism (rare) — sustained maternal hypercalcaemia suppresses fetal PTH
Clinical Features
Neonatal hypocalcemia manifests as a continuum of neuromuscular hyperexcitability, reflecting the fundamental role of ionised calcium in stabilising voltage-gated ion channels. When ionised calcium falls below 1.0 mmol/L, the threshold for spontaneous neuronal and muscle depolarisation is lowered — producing the characteristic picture of irritability and increased excitability even at rest. The severity of symptoms correlates imperfectly with serum calcium levels, partly because the ionised fraction (rather than total calcium) is the physiologically relevant variable, and partly because the rate of fall matters as much as the absolute value. It is also important to recognise that the clinical signs of neonatal hypocalcemia are non-specific and overlap substantially with neonatal hypoglycemia, sepsis, and hypoxic-ischaemic encephalopathy — making biochemical confirmation essential whenever a neonate presents with jitteriness, apnoea, or seizures. One distinctive feature is that hypocalcaemic seizures tend to be multifocal and clonic rather than the subtle eye-fluttering of hypoglycaemic encephalopathy, though this distinction is unreliable without EEG.
Asymptomatic hypocalcemia is common, particularly in early-onset disease among premature neonates. It is detected only by routine monitoring of at-risk groups.
Symptomatic hypocalcemia features:
- Jitteriness and tremors — high-frequency, low-amplitude limb trembling, often stimulus-sensitive; the most common presentation
- Irritability and high-pitched cry — neuromuscular irritability without obvious trigger
- Hypotonia — can paradoxically coexist with jitteriness; reflects global neuromuscular dysregulation
- Apnoea — particularly in preterm neonates; calcium is required for diaphragmatic contractility
- Seizures — multifocal clonic or tonic; a serious sign requiring urgent treatment
- Laryngospasm — stridor and sudden cyanosis due to vocal cord spasm; rare but immediately life-threatening
- Carpopedal spasm — wrist flexion with metacarpophalangeal extension and pedal spasm (Trousseau-like); seen mainly in older infants, uncommon in the first week
- ECG changes — prolonged corrected QT interval (QTc >0.45 seconds) is a reliable early marker of hypocalcemia, visible before symptoms; ST-segment changes may also occur
Chvostek's sign (twitching of the facial muscles on tapping the facial nerve anterior to the ear) is positive in neonatal hypocalcemia but is also positive in 10–25% of normal neonates, limiting its specificity.