Page 14 of 20
PE21.5 | Juvenile Idiopathic Arthritis — SDL Guide (Part 2)
Complications: Uveitis and Macrophage Activation Syndrome
Two complications of JIA demand particular attention because they are treatable if detected early but catastrophic if missed: chronic anterior uveitis and macrophage activation syndrome (MAS).
Chronic Anterior Uveitis
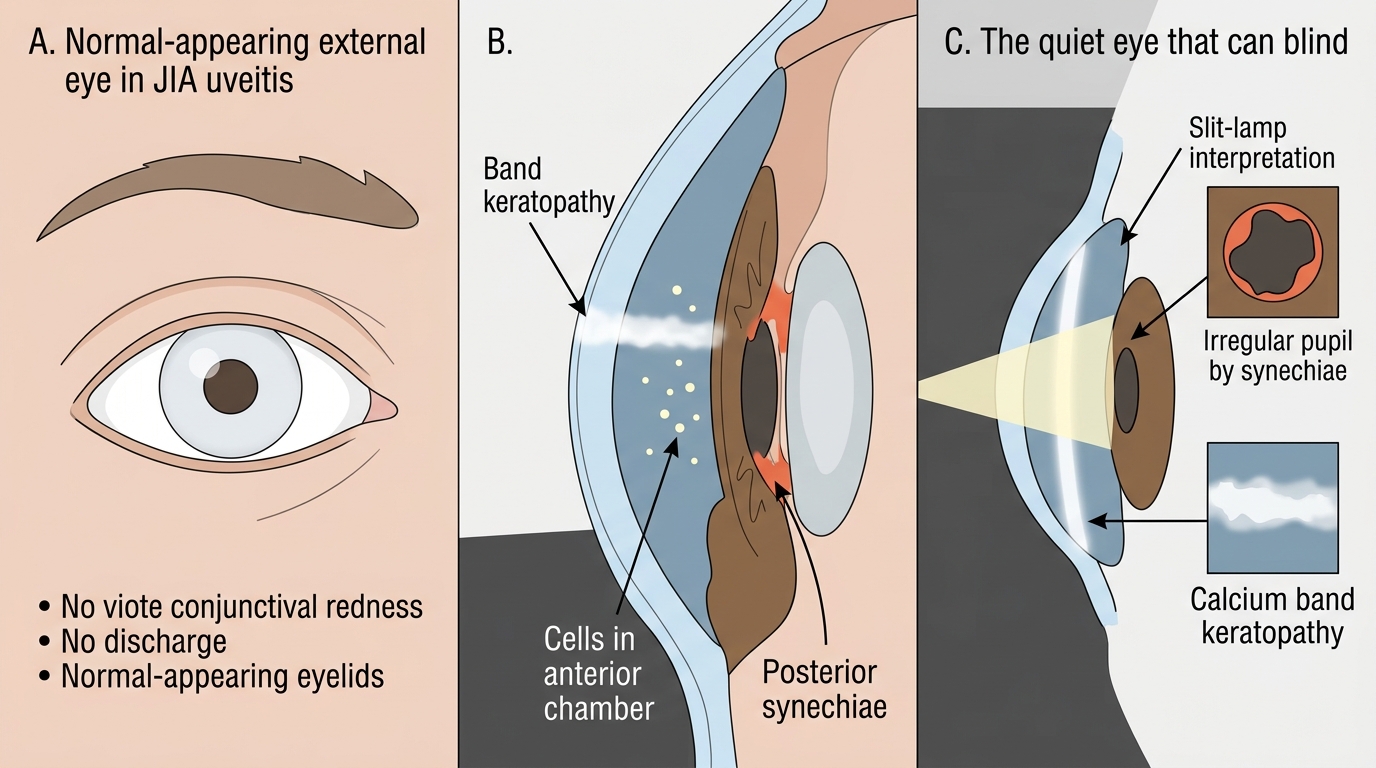
Uveitis is the most important extra-articular complication of JIA, occurring in approximately 10–20% of all JIA cases but in up to 30% of oligoarticular ANA-positive patients. The critical clinical point — one that has cost many children their vision — is that JIA-associated uveitis is entirely asymptomatic in the majority of cases. There is no redness, no photophobia, and no blurred vision until the disease is advanced. By the time symptoms appear, the damage is done: band keratopathy (calcium deposits across the cornea), posterior synechiae (adhesions between the iris and lens), cataract, glaucoma, and ultimately blindness may have developed.
This is why routine, regular slit-lamp biomicroscopy is mandatory, not optional, for all JIA patients. Screening frequency is determined by risk stratification:
• Highest risk (oligoarticular or polyarticular JIA + ANA-positive + onset <6 years): slit-lamp every 3 months
• Intermediate risk: every 6 months
• Lowest risk (ERA, systemic JIA, RF-positive polyarticular): every 12 months (ERA and systemic JIA have a lower uveitis risk)
Treatment of active uveitis involves topical corticosteroids and mydriatics to prevent synechiae, with systemic methotrexate and biologics (adalimumab is particularly effective) for steroid-dependent or refractory cases.
⚑ AI image — pending faculty review (auto-QA score 6/10; best of 3 attempts)
Slit-Lamp Findings of Anterior Uveitis in JIA
Macrophage Activation Syndrome (MAS)
MAS is a life-threatening complication of systemic JIA, occurring in 10–15% of systemic JIA patients at some point in their disease course. It is a secondary haemophagocytic lymphohistiocytosis (HLH) — an uncontrolled activation of macrophages and T lymphocytes driven by excess IL-1β and interferon-gamma.
Clinically, MAS presents as sudden clinical deterioration in a child with systemic JIA: high unremitting fever, hepatosplenomegaly, lymphadenopathy, and a bleeding tendency (petechiae, mucosal bleeding). The paradoxical laboratory hallmark is falling ESR (because fibrinogen — an acute-phase reactant — is consumed by phagocytosis) alongside rising ferritin (often >500 ng/mL; in severe MAS may exceed 10,000 ng/mL). Cytopenias (falling WBC, haemoglobin, and platelet count despite active inflammation) and elevated liver enzymes round out the picture. Bone marrow biopsy shows haemophagocytosis — macrophages engulfing red cells, white cells, and platelets.
MAS is a haematological emergency. Treatment involves high-dose corticosteroids, cyclosporin A, and increasingly anakinra (IL-1 inhibitor) or etoposide in refractory cases.
CLINICAL PEARL
The quiet eye that blinds: the asymptomatic uveitis trap. The most dangerous and most preventable complication of JIA is chronic anterior uveitis in an ANA-positive oligoarticular child who has absolutely no eye complaints. The eye looks normal from the outside. The child does not complain. The parent does not worry. Yet slit-lamp examination reveals cells in the anterior chamber, posterior synechiae, and early band keratopathy — changes that, untreated, lead to irreversible blindness. Every JIA patient, regardless of whether they have eye symptoms, must be enrolled in an ophthalmology surveillance programme. When the first symptom appears (blurred vision, photophobia, red eye), the disease is advanced. Risk-stratify: ANA-positive oligoarticular with onset <6 years = every 3 months without fail.
Diagnosis and Investigation of JIA
JIA is a clinical diagnosis of exclusion — there is no single diagnostic test that confirms it, and no biomarker that is both sensitive and specific. The diagnosis is made when a child meets the ILAR criteria (arthritis onset <16 years, duration ≥6 weeks) AND other identifiable causes of childhood arthritis have been systematically excluded. This exclusionary approach is non-negotiable: infections, malignancies, and other connective tissue diseases can all mimic JIA, and initiating immunosuppression before excluding these is dangerous.
History and Examination
Key clinical clues: age at onset, number and distribution of affected joints, presence of systemic features (fever pattern — is it quotidian?), rash, family history of psoriasis or inflammatory bowel disease (for ERA/psoriatic), enthesitis (heel pain, plantar fasciitis), uveitis symptoms, and history of recent infection.
Investigations — Routine
| Investigation | Purpose | Interpretation in JIA |
|---|---|---|
| Full blood count (CBC) | Rule out leukaemia; assess anaemia | Normocytic anaemia of chronic disease; thrombocytosis (reactive); leucocytosis in systemic JIA |
| ESR and CRP | Disease activity markers | Elevated in systemic > polyarticular > oligoarticular |
| ANA (antinuclear antibody) | Risk-stratify uveitis | Positive in 70–80% oligoarticular; NOT diagnostic of JIA |
| Rheumatoid Factor (RF) | Subtype polyarticular | RF+ on two occasions ≥3 months apart = RF-positive subtype |
| HLA-B27 | Support ERA diagnosis | Positive in ~80% ERA; NOT diagnostic alone |
| Anti-CCP antibodies | RF-positive subtype | More specific than RF for polyarticular RF+ subtype |
| Synovial fluid analysis | Rule out septic arthritis | JIA: inflammatory (WBC 5,000–80,000), sterile; septic: WBC >100,000, bacteria |
| X-ray of affected joints | Baseline; assess erosions | Early: periarticular osteoporosis, soft-tissue swelling; late: joint space narrowing, erosions |
| MRI joints | Sensitive for early synovitis | Detects synovial enhancement, effusion, early cartilage loss before X-ray changes |
| Slit-lamp examination | Screen for uveitis | Must be performed at diagnosis and on scheduled intervals regardless of symptoms |
Investigations to Exclude Other Diagnoses
Leukaemia must be actively excluded: if the child has bone pain (rather than joint swelling), night sweats, anaemia disproportionate to inflammation, or thrombocytopenia, perform peripheral blood smear and bone marrow biopsy before starting any immunosuppression. Do NOT mistake leukaemic arthralgia for JIA.
Septic arthritis: if any single hot, acutely tender joint is present with high fever and elevated inflammatory markers, joint aspiration and blood culture must precede any anti-inflammatory treatment.
Reactive arthritis: documented preceding infection, HLA-B27 status, and typically self-limiting course within 6 weeks distinguishes it from JIA.
Diagnostic approach summary:
1. Clinical diagnosis (ILAR criteria met)
2. Baseline bloods: CBC, ESR, CRP, ANA, RF, HLA-B27 if ERA suspected
3. Imaging: X-ray baseline; MRI for early/unclear cases
4. Slit-lamp: at diagnosis, then per risk-stratified schedule
5. Exclude infection (blood culture, joint aspiration if indicated) and malignancy
SELF-CHECK
A 12-year-old boy presents with bilateral heel pain (insertional) and pain in the right sacroiliac joint for 4 months. He is HLA-B27 positive. RF and ANA are negative. Which JIA subtype is most likely?
A. Oligoarticular JIA
B. Polyarticular RF-negative JIA
C. Enthesitis-related arthritis (ERA)
D. Systemic JIA
Reveal Answer
Answer: C. Enthesitis-related arthritis (ERA)
Enthesitis-related arthritis (ERA) is defined by arthritis AND/OR enthesitis (inflammation at tendon/ligament insertions such as the Achilles heel and plantar fascia) in a child who is HLA-B27 positive. It predominantly affects older boys (>8 years), involves large lower-limb joints and axial structures (sacroiliac joints), and may progress to ankylosing spondylitis. RF and ANA are negative. Oligoarticular JIA affects young girls with large joints and no enthesitis. Systemic JIA requires quotidian fever plus systemic features.
Management: NSAIDs, DMARDs, and Biologics
The management of JIA follows a stepwise, subtype-guided approach with the primary goals of suppressing inflammation, preserving joint function, preventing complications (particularly uveitis-related blindness and growth failure), and allowing the child to participate in normal childhood activities. Modern treatment with early aggressive therapy has transformed the outlook — most children achieve remission or low disease activity.
Step 1 — NSAIDs (Non-Steroidal Anti-Inflammatory Drugs)
NSAIDs are the first-line agents for pain and stiffness in mild oligoarticular disease. Naproxen (10–20 mg/kg/day in two divided doses) and ibuprofen (30–40 mg/kg/day in three to four divided doses) are the most used in paediatric practice. Naproxen has been associated with naproxen-induced pseudoporphyria (photosensitive skin fragility) in fair-skinned children — an important side effect to know. NSAIDs provide symptomatic relief but do NOT prevent joint erosions or uveitis; they are a bridge, not a disease-modifying treatment.
Step 2 — Intra-articular Corticosteroids (IACS)
Triamcinolone hexacetonide intra-articular injection is highly effective for oligoarticular JIA with 1–2 affected joints and is a standard treatment at this step. A single injection can provide disease control for months. Triamcinolone hexacetonide is preferred over triamcinolone acetonide due to its longer duration. Systemic corticosteroids are used judiciously — short courses for bridging or systemic JIA flares — but long-term oral steroids are avoided because of growth suppression, osteoporosis, cushingoid effects, and cataract.
Step 3 — Conventional DMARDs (Disease-Modifying Antirheumatic Drugs)
Methotrexate (MTX) is the anchor DMARD and the most widely used disease-modifying agent in JIA. It is indicated when NSAIDs alone are insufficient (extended oligoarticular, polyarticular, or uveitis not responding to topical therapy). Dosage: 10–15 mg/m²/week (oral or subcutaneous), with folic acid supplementation (1 mg/day) to reduce mucosal and hepatic side effects. Clinical response typically requires 4–6 weeks, with maximal benefit at 3–6 months. Monitoring: CBC, LFTs regularly. MTX is also effective for JIA-associated uveitis, particularly when combined with biologics.
Other conventional DMARDs sometimes used include leflunomide and sulfasalazine (particularly for ERA).
Step 4 — Biologics
Biologics are indicated when conventional DMARDs fail or in high-risk/severe disease:
- TNF-α inhibitors — Etanercept (TNF receptor fusion protein) and adalimumab (monoclonal anti-TNF antibody) are the most established biologics in JIA. Adalimumab is particularly effective for JIA-associated uveitis. Both are approved for polyarticular and oligoarticular JIA refractory to MTX. Screening for tuberculosis (TST/IGRA) is mandatory before starting any biologic due to reactivation risk.
- IL-6 inhibitor — Tocilizumab: approved for systemic JIA and polyarticular JIA. Highly effective for systemic features and MAS prevention in systemic JIA.
- IL-1 inhibitors — Anakinra and Canakinumab: canakinumab is approved for systemic JIA; anakinra for MAS acute management. These target the IL-1β pathway that is central to systemic JIA pathogenesis.
- CTLA4-Ig — Abatacept: a co-stimulation blocker that prevents T-cell activation; approved for polyarticular JIA refractory to other biologics.
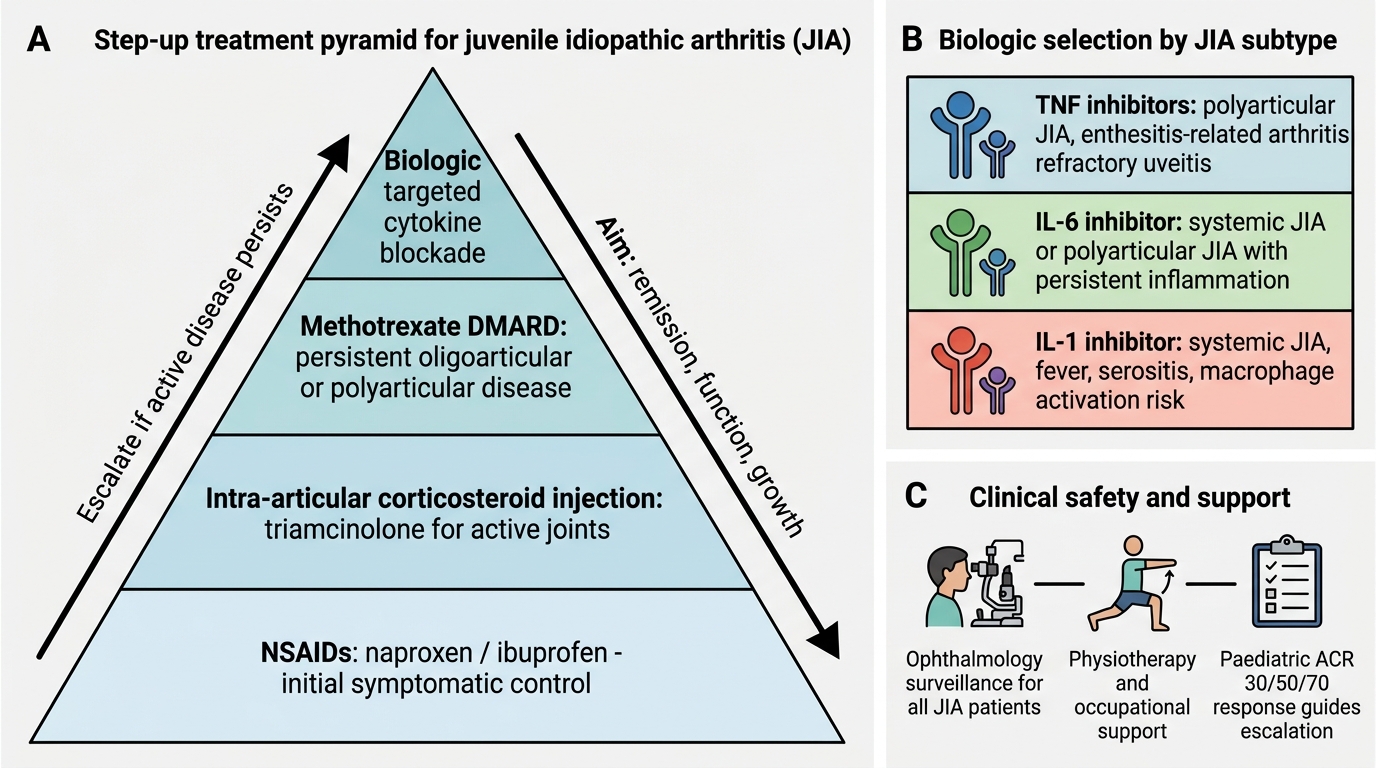
Step-up Treatment Pyramid for JIA
Ophthalmology Monitoring — Non-Negotiable
All JIA patients must be enrolled in ophthalmology surveillance regardless of whether they have eye symptoms. Frequency depends on risk stratification (see Complications section). Slit-lamp examination is the standard; ophthalmoscopy does not detect anterior uveitis.
Physiotherapy and Rehabilitation
Physiotherapy is an essential and often underemphasised component: range-of-motion exercises maintain joint flexibility, muscle strengthening prevents atrophy, and hydrotherapy is particularly well tolerated in children. Occupational therapy assists with school aids and daily activities. Splinting may correct or prevent joint deformities.
Monitoring of Disease Activity
The ACR/PRINTO paediatric response criteria (Paediatric ACR 30/50/70) define clinical improvement as ≥30%/50%/70% improvement across a core set of variables: physician global, parent/patient global, number of active joints, joints with limited range of motion, ESR/CRP, and functional ability. These thresholds guide treatment escalation decisions.
SELF-CHECK
A 10-year-old girl with polyarticular RF-negative JIA has had inadequate response to naproxen and intra-articular triamcinolone over 4 months. The next appropriate step in management is:
A. Long-term oral prednisolone 1 mg/kg/day
B. Methotrexate 10–15 mg/m²/week with folic acid
C. Adalimumab as first-line biologic
D. Sulfasalazine as preferred DMARD for RF-negative JIA
Reveal Answer
Answer: B. Methotrexate 10–15 mg/m²/week with folic acid
Methotrexate is the anchor conventional DMARD in JIA and is the standard next step when NSAIDs and intra-articular corticosteroids are insufficient. Dosage is 10–15 mg/m²/week with folic acid supplementation to reduce toxicity. Long-term oral corticosteroids are avoided in children due to growth suppression and metabolic effects. Biologics (adalimumab, etanercept) are reserved for failure of or contraindication to conventional DMARDs — they are not first-line. Sulfasalazine is particularly used for ERA rather than RF-negative polyarticular JIA.
Self-Assessment
Use the following case scenario to test your integrated understanding of JIA across all five arc steps: clinical presentation and subtype recognition, pathophysiology, investigation, management, and complication surveillance. Work through each question systematically before reading the model answers. Self-directed clinical reasoning — not passive recall — is the goal of this section.
Case: A 6-year-old girl is referred with a 5-month history of swelling and stiffness in her right knee and left ankle. Morning stiffness lasts about 45 minutes. There is no fever. Examination confirms warm, non-tender joint effusions bilaterally. No rash, no lymphadenopathy, no hepatosplenomegaly. Blood results: CBC — mild normocytic anaemia (Hb 10.8 g/dL); ESR 42 mm/h; CRP 18 mg/L; ANA 1:160 (positive); RF negative; HLA-B27 negative. X-rays show periarticular soft-tissue swelling with no erosions.
Questions to work through:
- Identify the subtype. She has arthritis for >6 weeks, onset <16 years, 2 joints (≤4 in first 6 months), ANA-positive, RF-negative, HLA-B27 negative, no systemic features. → This is oligoarticular JIA, persistent subtype.
- What is the single most important next investigation and why? → Slit-lamp ophthalmology examination. ANA-positive oligoarticular JIA with onset before age 7 is the highest-risk category for chronic anterior uveitis. The examination is mandatory at diagnosis and then every 3 months, regardless of the absence of eye symptoms.
- Outline initial management. → NSAIDs (naproxen or ibuprofen, weight-based dosing) for symptomatic relief + intra-articular triamcinolone hexacetonide to both affected joints. If inadequate response at 4–6 months, escalate to methotrexate (10–15 mg/m²/week with folic acid).
- What complication would change your management urgently? → Active uveitis on slit-lamp examination. Requires topical corticosteroids + mydriatics acutely; if persistent/refractory, add systemic methotrexate ± adalimumab.
- What growth-related complication might develop if this knee remains inflamed for years? → Leg length discrepancy — chronic hyperaemia from knee synovitis accelerates growth plate activity on the affected side, causing overgrowth of the inflamed limb. Conversely, systemic inflammation and steroid use can cause generalised growth failure.
- Name two systemic JIA-specific complications not present in this case. → (a) Macrophage activation syndrome (MAS) — falling ESR, rising ferritin, cytopenias, haemophagocytosis; (b) Serositis (pericarditis, pleuritis) — a defining criterion for systemic JIA that requires systemic corticosteroids or IL-1 inhibitors.
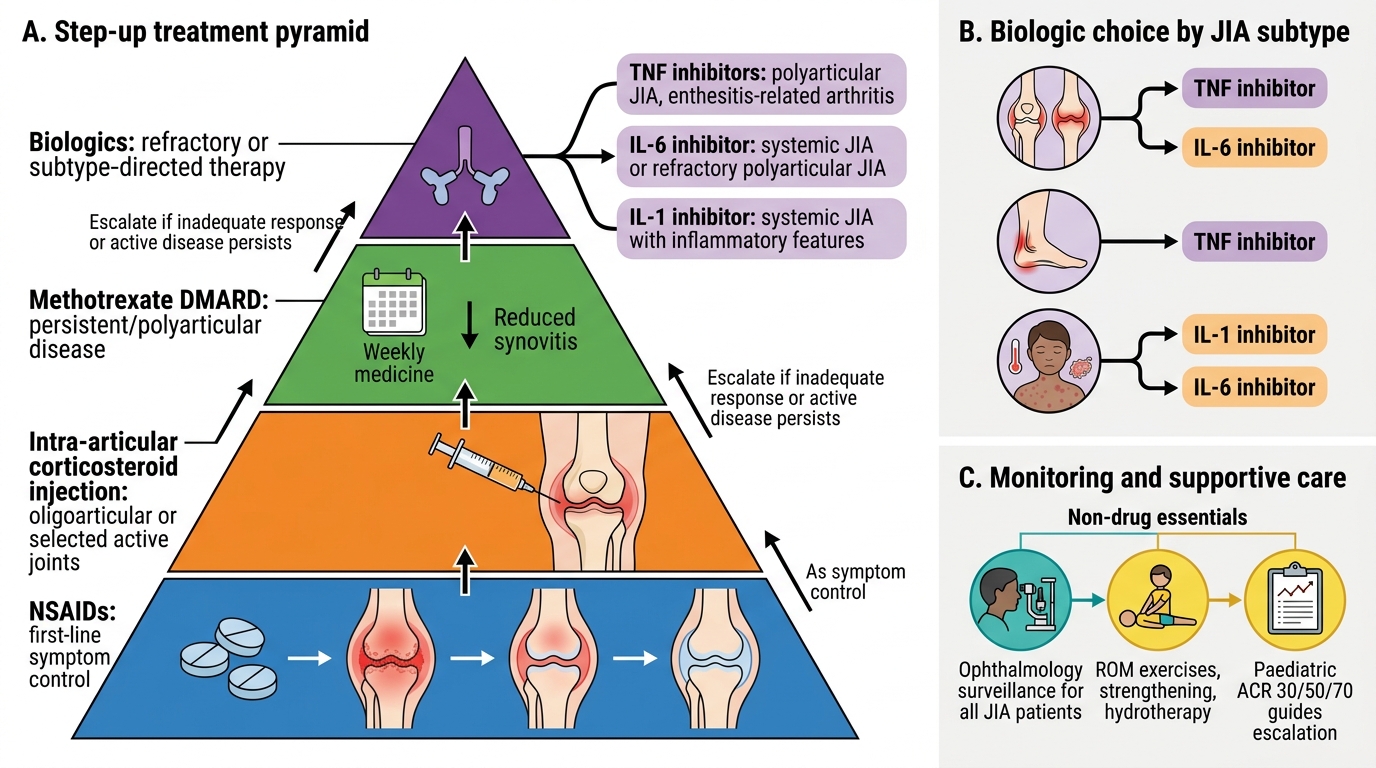
Step-up Management of Juvenile Idiopathic Arthritis