Page 27 of 42
PE23.14 | Fulminant Hepatic Failure — SDL Guide (Part 2)
Pathophysiology of Hepatic Encephalopathy
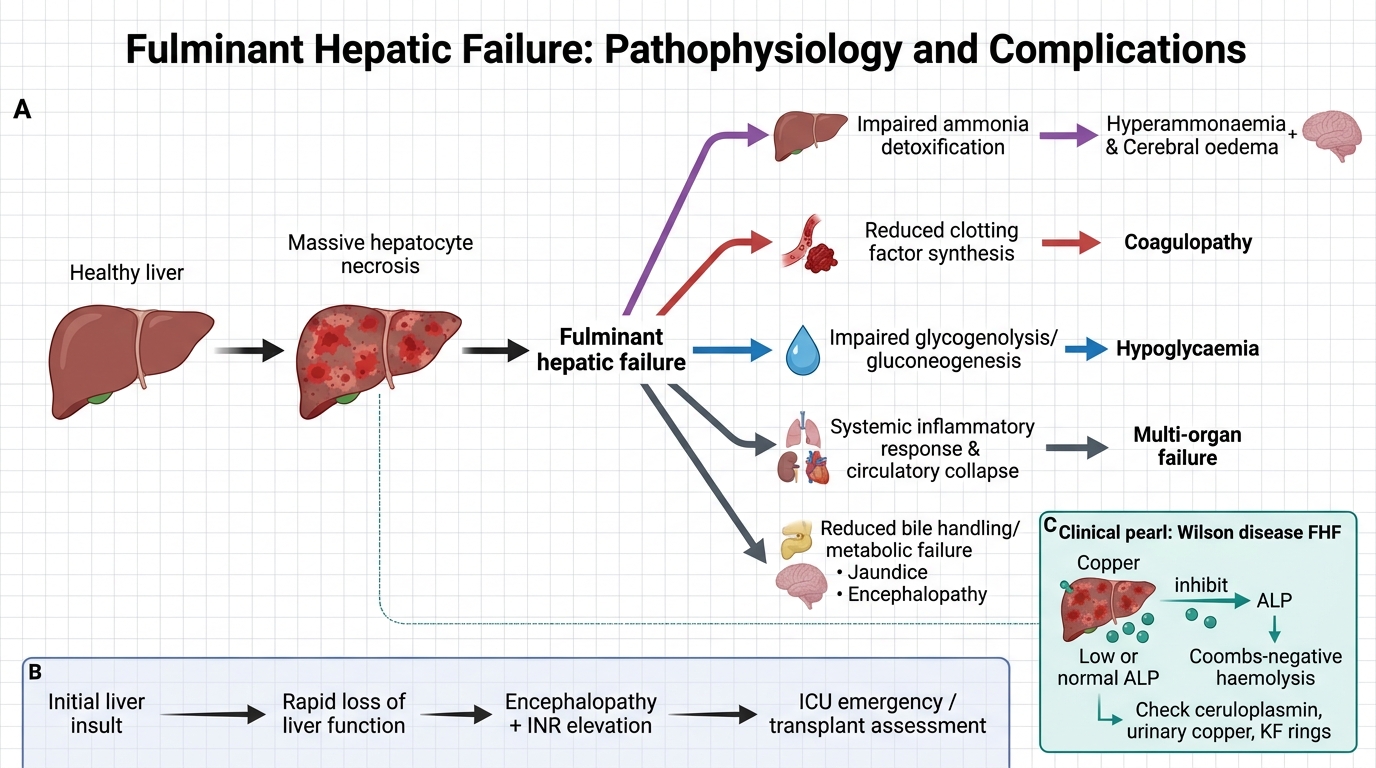
The pathophysiology of FHF involves a catastrophic cascade initiated by massive hepatocyte loss, which simultaneously disrupts all three major functions of the liver: metabolic detoxification, protein synthesis, and biliary secretion. Understanding this cascade helps rationalise every management intervention.
1. Ammonia accumulation and cerebral oedema:
The liver normally converts ammonia (produced by gut bacteria from protein breakdown and from enteric urea) to urea via the urea cycle. In FHF, this capacity is lost. Hyperammonaemia → astrocyte swelling (astrocytes convert ammonia to glutamine, which acts as an osmole) → cytotoxic cerebral oedema. Elevated intracranial pressure (ICP) leads to herniation and death. Astrocyte swelling also disrupts GABAergic and glutamatergic neurotransmission, contributing to encephalopathy.
2. Coagulopathy:
All clotting factors except Factor VIII are synthesised in the liver. Massive hepatocyte loss → reduced synthesis of Factors II, V, VII, IX, X, and fibrinogen → prolonged PT/INR. Additionally, thrombocytopenia may result from hypersplenism or disseminated intravascular coagulation (DIC), worsening bleeding tendency.
3. Hypoglycaemia:
Glycolysis and gluconeogenesis are both hepatic-dependent. Massive liver failure depletes glycogen and impairs gluconeogenesis → severe hypoglycaemia, which compounds encephalopathy by causing neuronal energy failure.
4. Systemic inflammatory response:
Massive hepatocyte necrosis releases damage-associated molecular patterns (DAMPs), triggering a systemic inflammatory response. This predisposes to bacterial infections (spontaneous bacterial peritonitis, pneumonia, urinary sepsis), which worsen HE and coagulopathy.
5. Renal failure (hepatorenal syndrome):
Splanchnic vasodilation (from nitric oxide and other mediators) leads to effective arterial underfilling → renal vasoconstriction → acute kidney injury without intrinsic renal pathology. This is functional and reversible with liver recovery or transplant.
Pathophysiology of Fulminant Hepatic Failure
CLINICAL PEARL
The paradox of alkaline phosphatase in Wilson disease FHF: In most causes of FHF, alkaline phosphatase (ALP) is elevated. In Wilson disease presenting as FHF, ALP is paradoxically LOW or normal. This is because copper released from necrotic hepatocytes inhibits ALP. A low ALP in a child with FHF should immediately trigger a Wilson disease workup (ceruloplasmin, 24-hour urinary copper, slit-lamp examination for Kayser-Fleischer rings). The combination of low ALP + Coombs-negative haemolytic anaemia + FHF in a child > 3 years is virtually pathognomonic of Wilson disease.
Diagnosis and Investigation
Investigations in FHF serve two simultaneous purposes: confirming the diagnosis of FHF by establishing the severity of liver failure (INR, bilirubin, ammonia, blood glucose) and identifying the aetiology, which determines whether a specific therapy is available. Both goals must be pursued in parallel — waiting for aetiology results before starting supportive care is dangerous, but equally, starting treatment without a diagnostic workup means missing a Wilson disease or autoimmune hepatitis case that is treatable. All investigations should be drawn at first contact, before any blood products are given, as FFP will falsely normalise the INR. Investigations must be interpreted dynamically, because in FHF the clinical and biochemical picture can change within hours: a rising INR, a falling ALT in the context of worsening HE, or a rising creatinine each carry important prognostic and therapeutic implications and should trigger immediate reassessment of the management plan.
Establishing liver failure severity:
• Liver function tests (LFTs): Serum bilirubin (markedly elevated, predominantly direct/conjugated), ALT and AST (elevated, but may fall paradoxically as necrosis is complete and no viable hepatocytes remain — a falling ALT with worsening encephalopathy is ominous), serum albumin (low in prolonged FHF)
• Coagulation profile: PT and INR (INR >1.5 is required for FHF definition; INR >4 is associated with poor prognosis), APTT, fibrinogen (DIC screen)
• Blood glucose (fingerprick + formal — hypoglycaemia common and dangerous)
• Serum ammonia (elevated in HE; used for monitoring; not part of the formal definition)
• Renal function tests: Serum creatinine, urea, electrolytes (hyponatraemia common; hepatorenal syndrome)
• CBC: Anaemia, thrombocytopenia (hypersplenism, DIC), leucocytosis (infection)
• Arterial blood gas/lactate: Metabolic acidosis in severe FHF
Aetiology screen:
| Investigation | Aetiology screened |
|---|---|
| Anti-HAV IgM | Hepatitis A |
| Anti-HEV IgM, HEV RNA | Hepatitis E |
| HBsAg, anti-HBc IgM, HBeAg | Hepatitis B |
| EBV/CMV/HSV PCR or serology | Viral (other) |
| Serum ceruloplasmin (<20 mg/dL) + 24-h urine copper + slit-lamp | Wilson disease |
| ANA, anti-SMA, anti-LKM1, IgG | Autoimmune hepatitis |
| Paracetamol level | Paracetamol toxicity |
| Drug and toxin history | Drug-induced |
Imaging:
• Ultrasound abdomen: Liver size (enlargement early, shrinkage late and ominous), splenomegaly, ascites, portal vein patency, hepatic parenchymal echotexture. Does NOT diagnose the aetiology of FHF but assesses anatomy.
• CT/MRI brain: If raised ICP or herniation is suspected clinically.
Prognostic scoring:
The King's College Criteria (adapted for paediatrics by PALF group) and PELD/MELD-Na score are used to assess urgency for liver transplant listing. Key poor-prognosis markers in children: INR >4, bilirubin >300 µmol/L, young age, non-HAV/HEV aetiology, Grade III–IV HE.
SELF-CHECK
A 12-year-old child with FHF has a serum alkaline phosphatase (ALP) that is LOW, a Coombs-negative haemolytic anaemia, and a serum ceruloplasmin of 12 mg/dL. Which investigation is MOST important to arrange immediately after this finding?
A. Anti-HAV IgM
B. 24-hour urinary copper estimation and slit-lamp examination
C. Anti-HEV IgM
D. Paracetamol level
Reveal Answer
Answer: B. 24-hour urinary copper estimation and slit-lamp examination
Low ALP + Coombs-negative haemolytic anaemia + low ceruloplasmin in a child >5 years with FHF is the classical triad of Wilson disease presenting as acute liver failure. The 24-hour urinary copper and slit-lamp examination for Kayser-Fleischer rings are the next priority. This matters urgently because Wilson FHF requires emergency liver transplant evaluation — chelation alone (penicillamine) is insufficient and can worsen the acute presentation. HAV, HEV, and paracetamol screens are important but do not fit this specific clinical picture.
Management of FHF
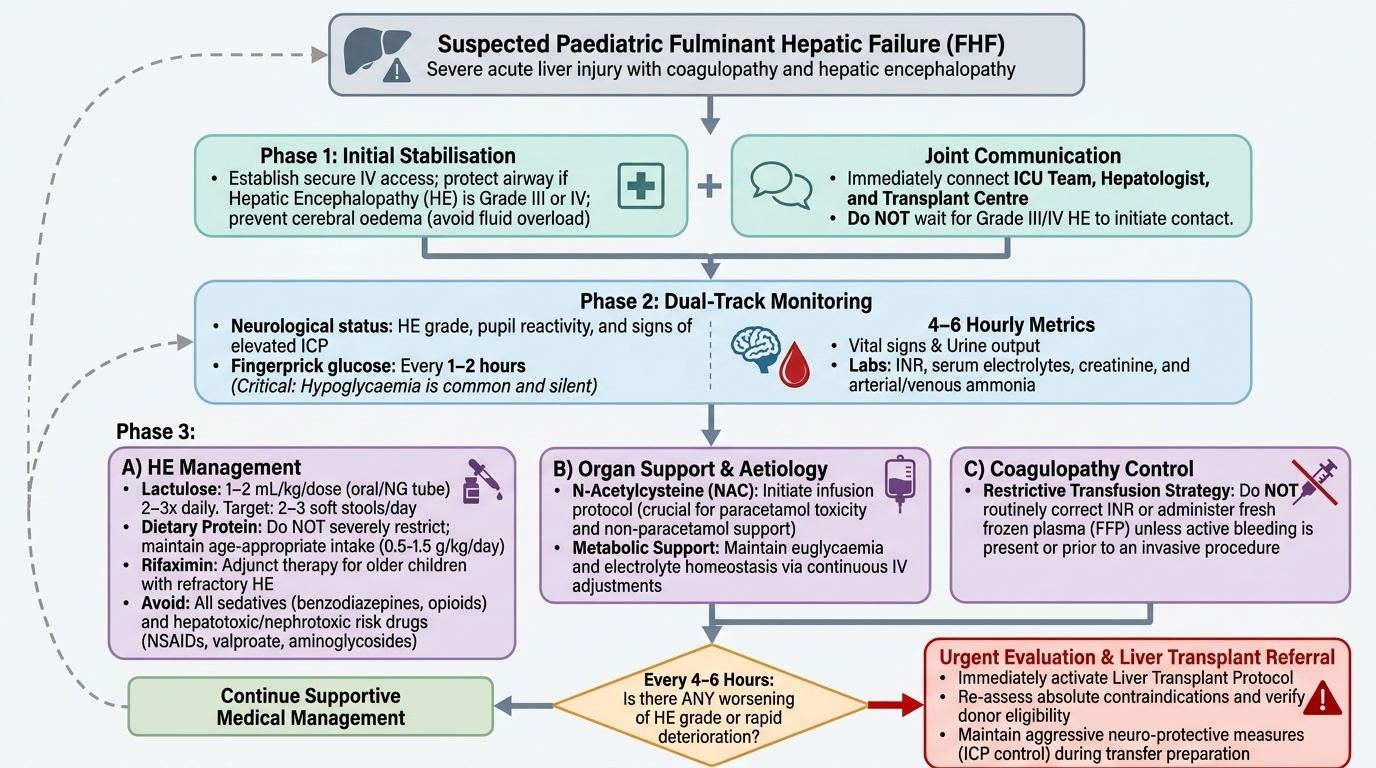
Management of FHF is simultaneously a battle on multiple fronts: preventing cerebral herniation from ammonia and oedema, correcting metabolic derangements (hypoglycaemia, electrolyte imbalance), controlling bleeding risk, preventing and treating infection, delivering any available aetiology-specific therapy, and determining whether the child needs liver transplant before the window of opportunity closes. No single intervention is curative except transplant for non-responders; the goal of medical management is to keep the child alive long enough for either spontaneous hepatic regeneration or transplant. Because FHF can deteriorate from Grade I to Grade IV HE within 24–48 hours, the management plan must be reviewed at least every 4–6 hours, and every worsening of HE grade should prompt reassessment of transplant candidacy. The ICU team, hepatologist, and transplant centre should be in communication from the time of diagnosis, not only when the child reaches Grade III–IV HE. The following priorities are presented in logical sequence but must often be pursued simultaneously.
Provided image
1. Monitoring:
• Continuous neurological assessment (HE grade, pupils, ICP signs)
• Fingerprick glucose every 1–2 hours (hypoglycaemia is common and silent)
• 4–6-hourly: vitals, urine output, INR, serum electrolytes, creatinine, ammonia
2. Hepatic encephalopathy management:
• Lactulose (orally or via nasogastric tube, 1–2 mL/kg/dose 2–3 times daily): reduces ammonia production by acidifying the colon (converting ammonia to non-absorbable ammonium) and accelerating faecal transit. Aim for 2–3 soft stools per day.
• Dietary protein: Do NOT severely restrict protein (worsens malnutrition and does not improve outcomes in children); provide age-appropriate protein (0.5–1.5 g/kg/day) with adjustment if ammonia levels do not improve.
• Rifaximin (in older children): non-absorbable antibiotic that reduces gut bacterial load; used as adjunct in refractory HE.
• Avoid hepatotoxic drugs: Stop valproate, NSAIDs, aminoglycosides (nephrotoxic in hepatorenal setting); use paracetamol only in reduced doses if analgesia is needed (paradoxically, low-dose paracetamol is safer than NSAIDs in liver disease).
• Avoid sedatives (benzodiazepines, opioids) as they precipitate or worsen HE.
3. Coagulopathy management:
• Do NOT routinely correct INR unless active bleeding or an invasive procedure is planned — coagulopathy also limits clot formation and a raised INR in FHF does not reflect true bleeding risk as well as in other contexts.
• For active bleeding: Fresh Frozen Plasma (FFP) 10–15 mL/kg; platelet concentrate if thrombocytopenic and bleeding; Vitamin K (phytomenadione) 1–5 mg IV (one dose — further doses are rarely helpful if the liver cannot synthesise factors).
• Avoid central line placement if possible; peripheral access preferred.
4. Hypoglycaemia correction:
• Maintain blood glucose 80–120 mg/dL with continuous IV dextrose (10% dextrose at maintenance rate; 12.5% or 15% via central line if needed).
5. N-Acetylcysteine (NAC):
• Indicated immediately for paracetamol overdose (most effective within 8–10 h; still beneficial up to 24 h). IV NAC: loading 150 mg/kg over 1 h, then 50 mg/kg over 4 h, then 100 mg/kg over 16 h.
• Also recommended in non-paracetamol FHF (PALF trial: NAC improved 1-year transplant-free survival in non-acetaminophen paediatric FHF, particularly Grades I–II HE). Dose as above for IV protocol.
6. Treating identified aetiology:
• Autoimmune hepatitis: Corticosteroids (dexamethasone IV) if liver biopsy confirms AIH and encephalopathy is not yet severe (Grade III–IV HE is a relative contraindication due to infection risk).
• Herpetic hepatitis: IV acyclovir.
• Wilson disease: Emergency liver transplant referral; do not start penicillamine in acute FHF (worsens acute presentation).
7. Infection prevention and treatment:
• Prophylactic antibiotics are NOT routinely recommended, but have a low threshold to culture (blood, urine, ascites) and start broad-spectrum antibiotics (ceftriaxone + metronidazole) if infection is suspected.
• Antifungal coverage (fluconazole) for those with prolonged ICU stay or broad-spectrum antibiotics.
8. Liver transplant evaluation:
Refer urgently to a transplant centre if: INR >4 despite optimal care, HE Grade III–IV, rising creatinine (hepatorenal syndrome), or identified Wilson disease. Early referral is critical — waiting until the child is moribund reduces transplant candidacy.
SELF-CHECK
An 8-year-old with FHF has a spontaneous nose bleed. His INR is 3.8 and platelets are 90,000/µL. He is Grade II HE. What is the MOST appropriate immediate action?
A. Give Vitamin K IV and FFP routinely to normalise the INR
B. Apply local pressure; give FFP 10-15 mL/kg and Vitamin K IV for the active bleeding, but do not correct INR prophylactically in the absence of active bleeding
C. Transfuse packed red blood cells immediately
D. Start prophylactic fresh frozen plasma to keep INR <2
Reveal Answer
Answer: B. Apply local pressure; give FFP 10-15 mL/kg and Vitamin K IV for the active bleeding, but do not correct INR prophylactically in the absence of active bleeding
Active bleeding in FHF is managed with FFP (10–15 mL/kg) and Vitamin K IV. However, prophylactic INR correction in the absence of active bleeding is NOT recommended — it does not reflect true haemostatic risk in FHF (the liver also makes anticoagulants and the net balance is complex), and routine FFP has risks (volume overload, infections). Vitamin K one dose is appropriate because a nutritional Vitamin K deficiency component may be present. Platelets of 90,000 with only nose bleed do not require platelet transfusion at this level unless actively bleeding from multiple sites.
Self-Assessment: FHF
Test your understanding with these integrated clinical scenarios.
Scenario 1: A 5-year-old girl develops jaundice 3 days after returning from a village fair. By day 7, she becomes drowsy and confused. Her INR is 2.4, ALT is 1800 U/L, anti-HAV IgM is positive. She is Grade II HE. What are your immediate management steps?
Key answer points: Diagnose FHF (viral hepatitis A aetiology, HE Grade II, INR >1.5, <8 weeks, no prior liver disease). Admit to ICU. Start lactulose, 10% dextrose infusion, monitor glucose 2-hourly, avoid sedatives. IV NAC (non-acetaminophen FHF). Arrange urgent transfer to liver transplant centre. Do NOT give steroids (viral aetiology). Monitor for cerebral oedema (Grade II moving to III = emergency).
Scenario 2: A 14-year-old presents with FHF. His ALP is low, he has haemolytic jaundice (Coombs-negative), and his ceruloplasmin is 8 mg/dL. His HE is Grade I. What specific diagnosis must be confirmed urgently, and what is the definitive treatment?
Key answer points: Wilson disease presenting as FHF (low ALP + Coombs-negative haemolytic anaemia + low ceruloplasmin). Confirm with 24-h urinary copper (markedly elevated) and slit-lamp for Kayser-Fleischer rings. Definitive treatment = liver transplant. Do NOT start penicillamine in acute FHF (worsens outcome). Supportive care while awaiting transplant.
Scenario 3: A 10-year-old with FHF is now Grade IV HE — unresponsive to all stimuli. His INR is 6.2, creatinine is rising. Parents ask: 'Is there any hope?' What do you tell them and what is the next step?
Key answer points: Grade IV HE with INR >4 and hepatorenal syndrome = very high mortality without transplant (medical management alone will not suffice). If transplant evaluation has not yet been initiated, this is an emergency referral. Discuss prognosis honestly and compassionately with family. Continue full ICU support (ventilation, pressors if needed, renal support) as bridge to transplant. Cerebral herniation prevention (head-up 30°, avoid hypotonic fluids, consider hypertonic saline for ICP).