Page 19 of 34
PE26.8 | Acute Lymphoblastic Leukemia — SDL Guide
Learning Objectives
- Describe the clinical presentation of ALL in children, including features of bone marrow failure (anaemia, thrombocytopenia, neutropenia), organomegaly, lymphadenopathy, bone pain, and extramedullary disease
- Explain the pathophysiology of clonal lymphoblast expansion, the classification into B-ALL and T-ALL, and the cytogenetic and clinical determinants of risk stratification
- Outline the diagnostic approach: CBC with peripheral smear, bone marrow examination (>25% blasts), immunophenotyping, cytogenetics, and staging investigations
- Describe the principles of risk-stratified ALL treatment phases (induction, consolidation, maintenance), CNS prophylaxis with intrathecal methotrexate, and the recognition and management of tumour lysis syndrome
INSTRUCTIONS
Acute lymphoblastic leukaemia is the most common malignancy of childhood, responsible for the majority of paediatric cancer-related morbidity and mortality in India. A final-year student must be able to recognise the clinical and haematological features that distinguish ALL from benign causes of thrombocytopenia or anaemia, initiate the correct diagnostic workup, and understand the treatment framework that has transformed ALL from a universally fatal disease into one with cure rates exceeding 85% in optimally treated patients.
References
- Ghai Essential Pediatrics, 9th ed., Ch. 16 (textbook)
- Nelson Textbook of Pediatrics, 21st ed., Ch. 526 (textbook)
- Indian Pediatric Oncology Group (InPOG) ALL-15 Protocol (guideline)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A 4-year-old girl is brought to the paediatric emergency with a two-week history of fever, pallor, and increasing lethargy. Her mother has noticed blood in her spit when brushing her teeth and several large bruises on her thighs. On examination: temperature 38.4°C, HR 140/min, pale conjunctivae, multiple cervical and axillary lymph nodes (each 1.5–2 cm, non-tender, non-matted), liver palpable 4 cm below costal margin, spleen 5 cm below costal margin. CBC: Hb 5.8 g/dL, WBC 94,000/µL, platelets 12,000/µL. Peripheral smear shows 78% blasts with scant cytoplasm, fine chromatin, and inconspicuous nucleoli. She is not just thrombocytopenic — something is filling her marrow and spreading through her body. What is it, and can she be cured?
WHY THIS MATTERS
Acute lymphoblastic leukaemia (ALL) is the single most common malignancy in children, accounting for approximately 30% of all childhood cancers and about 80% of childhood leukaemias. It is also one of paediatric oncology's greatest success stories: with modern multiagent chemotherapy and risk stratification, cure rates exceed 85–90% in high-income settings. In India, outcomes are improving with the adoption of standardised protocols through bodies such as the Indian Pediatric Oncology Group (InPOG). Every doctor practising in any setting must be able to recognise the clinical and haematological features of ALL, distinguish it from benign thrombocytopenia (such as ITP), and initiate appropriate urgent referral and supportive care.
RECALL
Activate these prior concepts before proceeding:
- Lymphopoiesis: B lymphocytes mature in the bone marrow from common lymphoid progenitors; T lymphocytes migrate to the thymus for maturation. Both originate from haematopoietic stem cells. ALL arises from the malignant transformation and arrest of either lineage at an early stage.
- CBC interpretation in children: Age-dependent normal values; in a 4-year-old, normal Hb ≈11–13 g/dL; WBC 6,000–17,000/µL; platelets 150,000–400,000/µL. Simultaneously low Hb, low platelets, and elevated WBC with blasts = marrow infiltration until proven otherwise.
- ITP vs leukaemia: The critical distinction — ITP presents with isolated thrombocytopenia in a well-looking child with no organomegaly; ALL presents with hepatosplenomegaly, lymphadenopathy, anaemia, and blasts. Never treat thrombocytopenia with steroids until this distinction is made.
- Tumour lysis syndrome: Rapid destruction of cancer cells releases intracellular contents: potassium (↑), phosphate (↑, which complexes calcium → ↓Ca), uric acid (↑ from purine nucleotide catabolism). The resulting metabolic emergency (hyperkalaemia, hyperphosphataemia, hyperuricaemia, hypocalcaemia) can cause acute renal failure, cardiac arrhythmia, and death.
Clinical Presentation of ALL
The clinical presentation of ALL is the direct consequence of two processes operating simultaneously: replacement of normal haematopoiesis by expanding malignant blasts (causing marrow failure symptoms), and infiltration of extramedullary organs by leukaemic cells (causing organomegaly, lymphadenopathy, and distant organ involvement). Understanding these two mechanisms allows you to predict and recognise essentially the entire symptom complex of ALL. The temporal onset is usually subacute — symptoms develop over days to weeks — and the child may appear surprisingly alert despite severe anaemia and thrombocytopenia. This subacute evolution means ALL is often misidentified initially as a viral illness, nutritional anaemia, or a benign bleeding disorder; a high index of suspicion in any child with persistent pallor, unexplained bruising, or prolonged fever is the key to early diagnosis.
Marrow failure symptoms arise when the expanding blast population physically crowds out normal erythroid, myeloid, and megakaryocytic progenitors:
• Anaemia (reduced red cell production): pallor, fatigue, tachycardia, breathlessness on exertion.
• Thrombocytopenia (reduced platelet production): petechiae, purpura, mucosal bleeding (gum, nose), menorrhagia in older girls.
• Neutropenia (reduced mature neutrophil production): recurrent or persistent fever, susceptibility to bacterial and fungal infections.
Extramedullary infiltration produces the features that distinguish ALL from ITP and aplastic anaemia:
• Hepatomegaly and splenomegaly — leukaemic blast infiltration of the liver and spleen; both organs can become massively enlarged, causing abdominal distension and discomfort.
• Lymphadenopathy — generalised, often bilateral cervical, axillary, and inguinal nodes; nodes are typically non-tender, firm, and non-matted.
• Bone pain and arthralgia — blast infiltration of the periosteum and metaphyseal bone is common in childhood ALL; a child presenting with limb pain and low-grade fever should have ALL considered, especially if CBC is abnormal. Bone pain that is out of proportion to findings is a classic presentation.
• CNS involvement (CNS-3 status): Leukaemic cells can penetrate the blood-brain barrier; presentation includes headache, vomiting (raised ICP), cranial nerve palsies (especially VII), and papilloedema. CNS disease requires intensification of treatment.
• Testicular involvement: Unilateral or bilateral painless testicular enlargement; the testis is a pharmacological sanctuary site because the blood-testis barrier limits drug penetration; suspected by palpation, confirmed by biopsy.
• Mediastinal mass: A distinguishing feature of T-ALL (thymic T lymphoblast origin); can compress the trachea and superior vena cava — presenting as stridor, orthopnoea, facial oedema, and engorged neck veins (SVC syndrome). A mediastinal mass in a child warrants urgent investigation before any procedure (including sedation or general anaesthesia, which can cause complete airway obstruction).
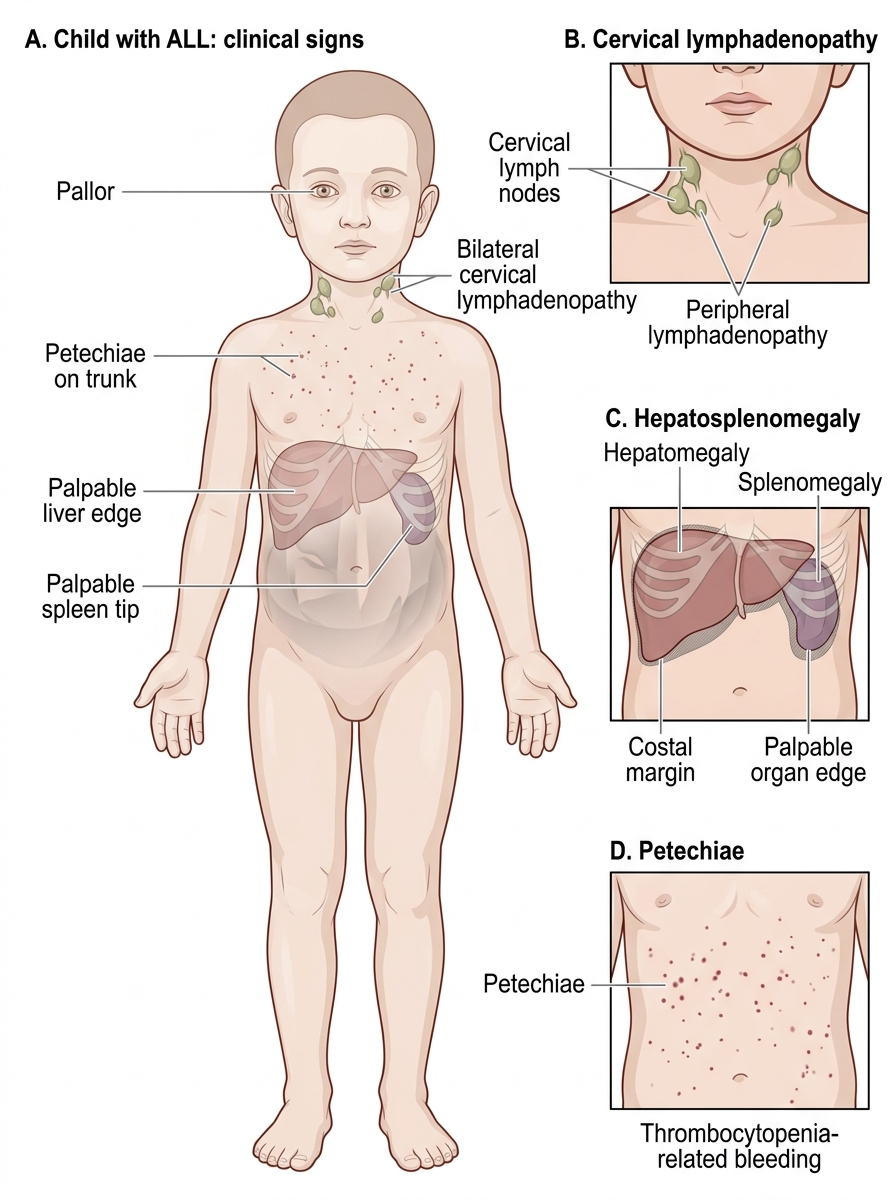
Clinical Signs of Childhood ALL
Pathophysiology, Aetiology and Classification
ALL arises from the clonal malignant transformation of a single lymphoid progenitor cell that acquires somatic mutations causing it to proliferate without the normal checkpoints and arrest of maturation at an early (immature, blast) stage. The result is an exponentially expanding clone of non-functional immature lymphocytes that fills the bone marrow, spills into the peripheral blood, and infiltrates extramedullary sites. Because the blasts cannot mature into functional immune cells, and because their expanding mass crowds out normal haematopoiesis, the patient develops both immune failure and bone marrow failure simultaneously. The arrested developmental stage of the leukaemic clone determines its immunophenotype — whether B-lineage or T-lineage — and profoundly influences the clinical presentation, the cytogenetic landscape, and the prognosis. Identifying lineage and cytogenetics is therefore not academic detail but the direct basis of risk stratification and treatment-intensity selection.
Classification by lineage is the first and most clinically important division:
• B-lineage ALL (B-ALL): Accounts for approximately 85% of childhood ALL. The transformed cell is a B-lymphoid progenitor arrested at the pre-B or early pre-B stage. Immunophenotype: TdT+, CD19+, CD10+ (also called CALLA, common ALL antigen), HLA-DR+. B-ALL encompasses a broad spectrum of cytogenetic subtypes with widely varying prognosis.
• T-lineage ALL (T-ALL): Approximately 15% of childhood ALL. The transformed cell is a T-lymphoid progenitor, originating in the thymus. Immunophenotype: TdT+, CD3+, CD7+, CD2+. T-ALL typically presents in older boys (>10 years), with a high WBC count, mediastinal mass, and CNS involvement — collectively defining a higher-risk clinical profile.
Cytogenetic and molecular determinants of prognosis are central to risk stratification:
• Hyperdiploidy (>50 chromosomes per blast cell): Found in ~25–30% of B-ALL; associated with excellent prognosis (~90% EFS); responds well to antimetabolite therapy.
• ETV6-RUNX1 (TEL-AML1) fusion: The most common chromosomal translocation in childhood ALL (~25%); excellent prognosis.
• BCR-ABL1 (Philadelphia chromosome, t(9;22)): Present in ~3–5% of childhood ALL (more common in adults); historically the worst prognosis; now treated with tyrosine kinase inhibitors (imatinib/dasatinib) plus chemotherapy with markedly improved outcomes.
• KMT2A (MLL) gene rearrangement: Characteristic of infant ALL (<1 year); extremely poor prognosis; requires intensive/SCT-based protocols.
• Hypodiploidy (<44 chromosomes): Associated with poor prognosis.
Risk factors for ALL include: Down syndrome (trisomy 21 — 10–20× increased ALL risk), prior exposure to ionising radiation or alkylating agents, Fanconi anaemia, Li-Fraumeni syndrome, and ataxia-telangiectasia. The majority of cases have no identifiable external risk factor.
| Feature | Standard Risk | High Risk |
|---|---|---|
| Age | 1–9 years | <1 year (infant) or ≥10 years |
| Initial WBC | <50,000/µL | ≥50,000/µL |
| Immunophenotype | B-ALL | T-ALL |
| Cytogenetics | Hyperdiploidy, TEL-AML1 | BCR-ABL1 (Ph+), KMT2A rearrangement |
| CNS disease | Absent | Present (CNS-2 or CNS-3) |
| Early treatment response (MRD) | Rapid clearance of blasts | Slow MRD clearance |
| Prognosis | ~90% 5-yr EFS | ~60–75% 5-yr EFS (varies by subtype) |
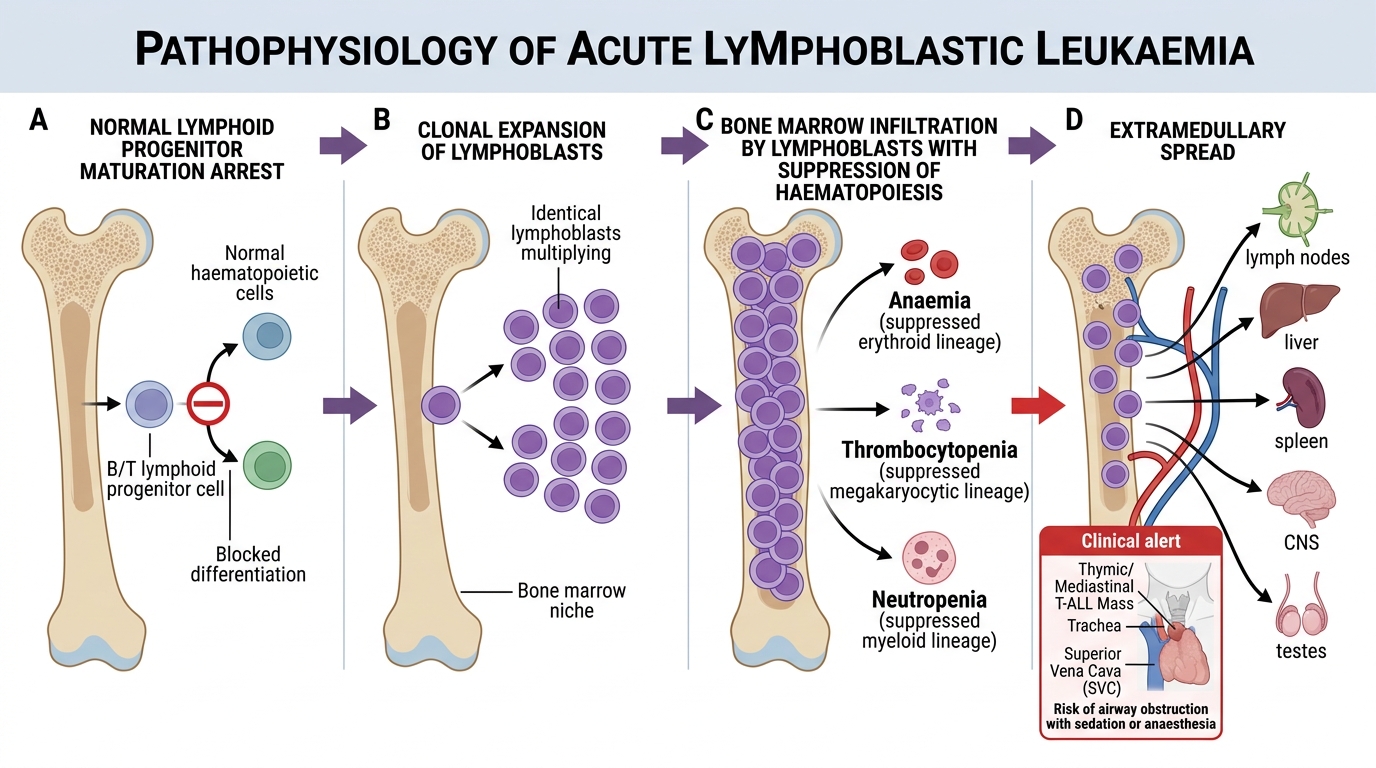
Pathogenesis and Spread of Acute Lymphoblastic Leukaemia
SELF-CHECK
A 13-year-old boy presents with superior vena cava (SVC) syndrome, a mediastinal mass on CXR, WBC 120,000/µL, and blasts positive for CD3 and TdT. Which type of ALL does he have, and what additional risk should you immediately anticipate?
A. B-ALL with Philadelphia chromosome; risk of cardiac arrhythmia
B. T-ALL with thymic origin; risk of complete airway obstruction under sedation or anaesthesia due to mediastinal compression
C. B-ALL with ETV6-RUNX1 fusion; standard risk disease
D. Infant ALL with KMT2A rearrangement; standard risk due to young age
Reveal Answer
Answer: B. T-ALL with thymic origin; risk of complete airway obstruction under sedation or anaesthesia due to mediastinal compression
CD3+, TdT+, mediastinal mass in an older boy = T-ALL (thymic origin). The mediastinal mass is the key danger: tracheal and SVC compression is present, and administering sedation or general anaesthesia can precipitate complete airway obstruction (tracheal collapse) — a life-threatening emergency. Any procedure in this scenario requires senior anaesthetic involvement and careful pre-operative assessment. T-ALL is also high-risk due to high WBC, mediastinal involvement, and tendency to CNS disease.
Diagnosis and Investigation
The diagnosis of ALL is confirmed by demonstration of ≥25% lymphoblasts in the bone marrow, supported by immunophenotyping and cytogenetic analysis. The clinical workup proceeds in a logical sequence from screening investigations to definitive diagnostic studies.
Peripheral blood CBC and smear: The CBC typically reveals some combination of anaemia, thrombocytopenia, and either leucocytosis (due to circulating blasts) or leucopenia (blasts trapped in marrow). The peripheral smear is essential: the presence of blasts — cells with a high nuclear-to-cytoplasmic ratio, immature (fine, stippled) chromatin, and occasional prominent nucleoli — on the smear is a haematological emergency requiring immediate referral to a paediatric haematology/oncology centre. Blasts on the smear (in a child with the above clinical picture) should NEVER be dismissed. The absence of blasts on the smear does not exclude ALL if the marrow is heavily infiltrated but the blasts have not yet spilled into blood (aleukemic presentation).
Bone marrow aspiration and biopsy: The definitive diagnostic procedure. The aspirate is examined under light microscopy; ≥25% lymphoblasts in the marrow (per WHO classification) establishes the diagnosis of ALL (the older 30% threshold may still be cited in some texts). The aspirate is simultaneously sent for immunophenotyping, cytogenetics, and molecular studies.
Immunophenotyping (flow cytometry): Antibody panels distinguish B-ALL (CD19+, CD10+/CALLA+, TdT+, HLA-DR+) from T-ALL (CD3+, CD7+, CD2+, TdT+, CD5+) and from AML (which may also show bone marrow blasts but expresses myeloid markers — CD13, CD33, CD117 — and lacks lymphoid markers). Immunophenotyping also detects aberrant lineage co-expression that can guide minimal residual disease (MRD) monitoring.
Cytogenetics and molecular studies: Conventional karyotype detects numerical abnormalities (hyperdiploidy, hypodiploidy) and structural rearrangements. Fluorescence in situ hybridisation (FISH) and RT-PCR detect specific translocations (BCR-ABL1, KMT2A) with higher sensitivity. These studies directly determine risk group assignment and treatment intensity.
Staging investigations: CSF examination (lumbar puncture) is performed after initiating induction to assess CNS involvement — ≥5 WBCs/µL with blasts = CNS-3 status. The LP timing in relation to chemotherapy requires careful planning to avoid complications. Chest X-ray or CT detects mediastinal mass (T-ALL). Testicular ultrasound or biopsy if clinical suspicion. Serum LDH and uric acid at baseline (markers of tumour burden and TLS risk).
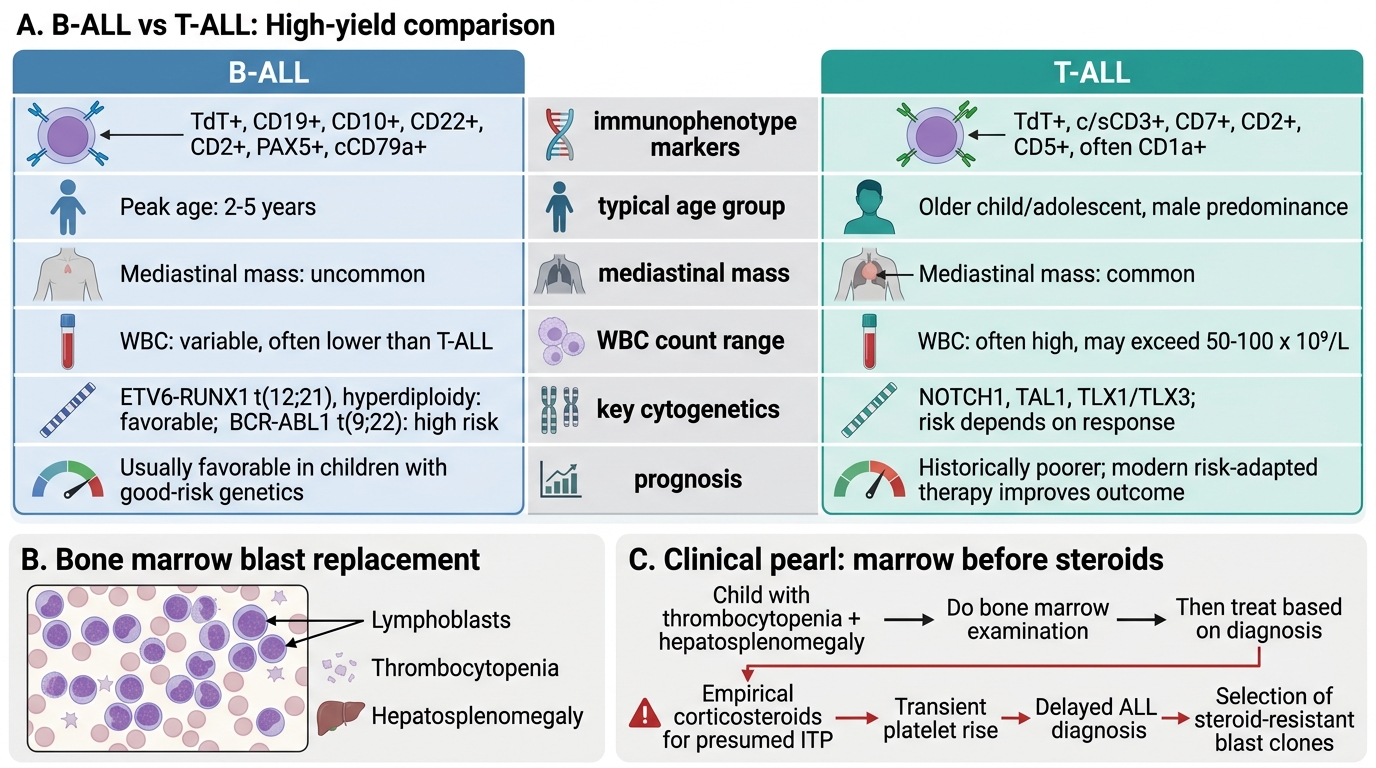
B-ALL vs T-ALL: High-Yield Comparison and Steroid Trap
CLINICAL PEARL
The hepatosplenomegaly trap — reiterated for ALL: Any child with thrombocytopenia AND hepatosplenomegaly must have a bone marrow examination before receiving corticosteroids. Steroids cause a transient improvement in platelet count in both ITP and ALL (because lymphoblasts express glucocorticoid receptors and are steroid-sensitive). Treating a child with ALL as if they have ITP with a steroid course delays the correct diagnosis by weeks, allows further leukaemic proliferation, and — critically — may select for steroid-resistant blast clones, worsening long-term prognosis. When in doubt: marrow before steroids.