Page 27 of 34
PE26.{10-15,17} | Hemato-Oncology Examination and Investigations — SDL Guide (Part 3)
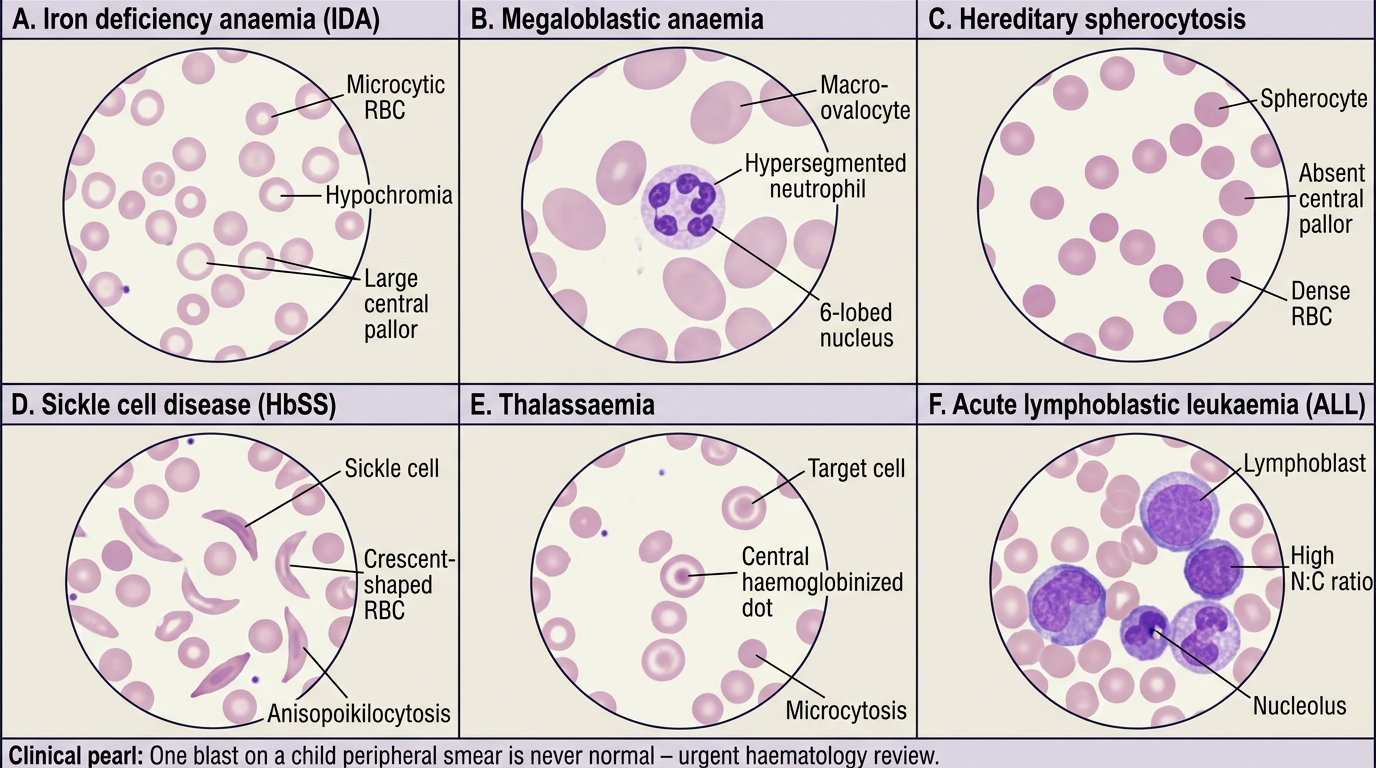
Interpretation of Findings: Peripheral Smear Morphology
The peripheral blood smear provides morphological information that no automated analyser can fully replicate: cell shape, surface pattern, inclusions, and blast morphology are visual findings that flow-cytometric haematology analysers cannot characterise. This is why a peripheral smear remains mandatory whenever the clinical picture does not fit the automated CBC result, whenever the analyser flags immature cells, whenever anaemia is unexplained, or whenever haematological malignancy is suspected. Systematic reading — size (microcytic/normocytic/macrocytic), colour (hypochromic/normochromic/hyperchromic), shape (poikilocytosis patterns), and inclusions for RBCs; then the WBC differential and morphology; then platelet morphology and estimate — prevents missed findings. Each morphological abnormality of the red cell carries a specific diagnostic implication, and the skill lies in recognising not just that a cell is abnormal but which abnormality it represents. For example, both spherocytes and sickle cells are abnormal red cell shapes, but they arise from entirely different pathological processes (membrane defect vs haemoglobin polymerisation) and lead to entirely different management. This section systematically maps each major morphological pattern to its clinical diagnosis and the confirmatory tests that follow.
Microcytic hypochromic pattern (MCV <72 fL, MCH low):
- IDA: pencil cells (elongated, thin), microcytes, prominent central pallor (>1/3 of cell diameter), anisocytosis (elevated RDW), poikilocytosis. Platelets may be elevated (reactive thrombocytosis).
- Thalassaemia trait: uniform microcytes, target cells, mild hypochromia, near-normal RDW. HbA2 elevated on electrophoresis confirms beta-thalassaemia trait.
- Thalassaemia major: severe microcytic hypochromic anaemia, target cells, nucleated RBCs (NRBCs), basophilic stippling, Cabot rings, marked anisocytosis.
- Anaemia of chronic disease: mild-to-moderate normocytic or microcytic anaemia, low reticulocyte count, no striking morphological abnormality.
Macrocytic pattern (MCV >100 fL):
- Megaloblastic anaemia (B12/folate deficiency): oval macrocytes (not just large round cells), hypersegmented neutrophils (≥5 nuclear lobes in ≥5% of neutrophils is the diagnostic criterion; ≥6 lobes in any neutrophil is strongly suggestive), macro-ovalocytes, giant platelets. This is the pathognomonic smear finding.
Specific morphological markers (high diagnostic specificity):
- Spherocytes: small, round, dense cells with absent central pallor, elevated MCHC. Seen in hereditary spherocytosis (osmotic fragility test positive) and autoimmune haemolytic anaemia (DAT positive). Distinguish from burned-out or fragment artefacts.
- Sickle cells (drepanocytes): elongated, crescent-shaped RBCs with pointed ends, in sickle cell disease (HbSS). Irreversible sickle cells appear even in the oxygenated blood sample of severe disease. Also seen in HbSC and HbS-thalassaemia.
- Target cells (codocytes): bull's-eye configuration, increased surface-to-volume ratio. In thalassaemia, HbC disease, liver disease (cholestasis), post-splenectomy, IDA (occasionally). The clinical context distinguishes.
- Blasts: large cells with high nuclear-to-cytoplasmic ratio, fine nuclear chromatin, prominent nucleoli, scant cytoplasm. Presence of even 1 blast on a well-prepared smear is abnormal and mandates urgent workup for acute leukaemia. Lymphoblasts (ALL) have scant agranular cytoplasm; myeloblasts (AML) may show Auer rods — pathognomonic of AML.
- Schistocytes (helmet cells, fragmented RBCs): microangiopathic haemolytic anaemia — DIC, HUS, TTP.
Peripheral Blood Smear Patterns in Anaemia and Leukaemia
CLINICAL PEARL
The one-blast rule: A single blast cell on a well-prepared peripheral smear is never normal in a child. Even if the WBC count is normal or only mildly elevated, one blast on smear is grounds for immediate repeat CBC, urgent haematology review, and bone marrow aspiration. The most dangerous error is to attribute a low TLC with blasts to a 'viral illness' — early leukaemia can present with leukopenia, and the blast percentage on BMA determines the diagnosis. Never reassure a family until a marrow examination has excluded leukaemia.
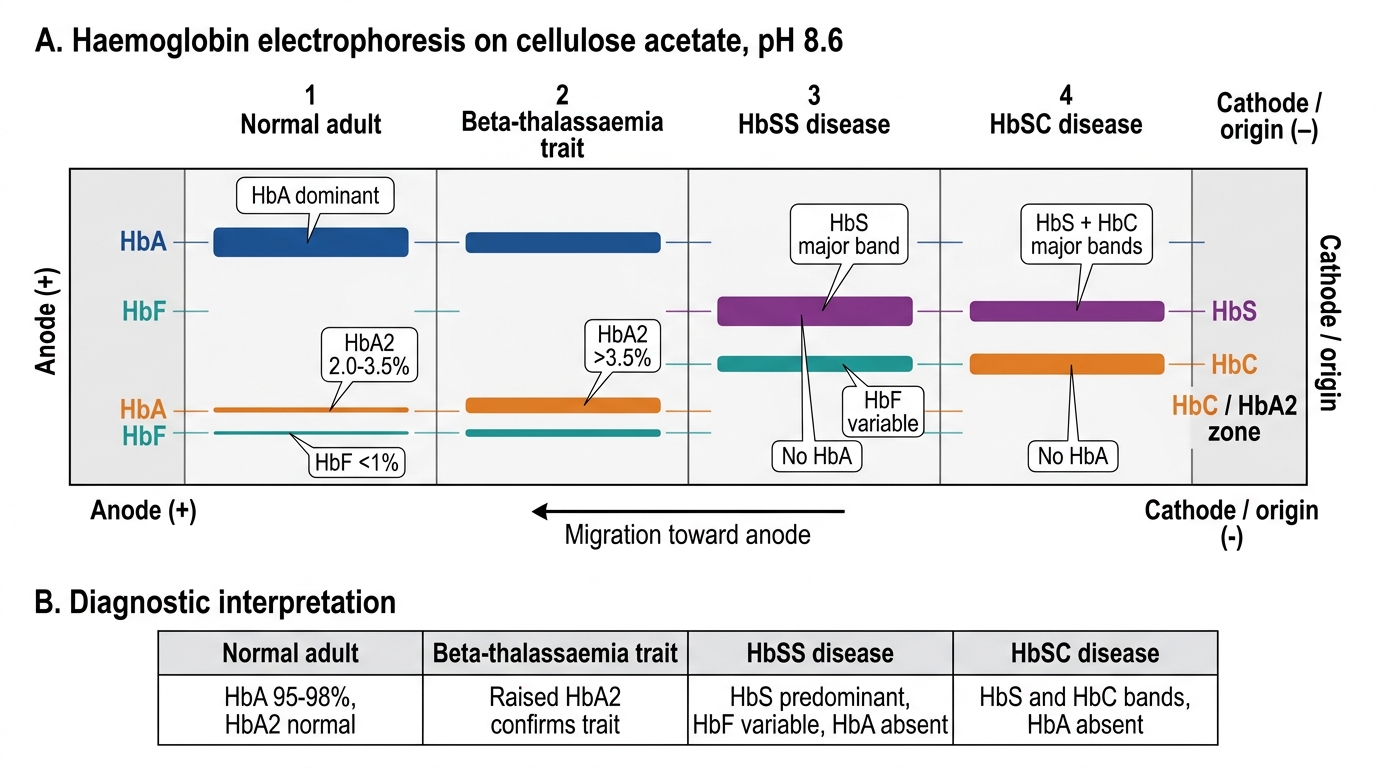
Interpretation of Findings: Haemoglobin Electrophoresis Report
Haemoglobin electrophoresis separates haemoglobin variants by their electrical charge at a defined pH and reports the percentage of each haemoglobin fraction. It is the definitive investigation for diagnosing haemoglobinopathies — a family of inherited disorders of haemoglobin structure or synthesis that together affect millions of Indian children. The principle relies on the fact that different haemoglobin variants (HbA, HbA2, HbF, HbS, HbC) carry different net electrical charges at a given pH, causing them to migrate at different speeds through a cellulose acetate membrane (alkaline pH 8.6) or a citrate agar gel (acid pH 6.2). Modern Indian referral laboratories increasingly use high-performance liquid chromatography (HPLC), which provides precise quantitative fractions, superior sensitivity for detecting trace variants, and a characteristic elution pattern for each haemoglobin type — making it both more accurate and faster than classical electrophoresis. Knowing which haemoglobin fractions to look for on the report, and what threshold for each fraction means clinically, is the competency this section builds. India carries the world's largest burden of thalassaemia carriers (approximately 3–4% of the general population are beta-thalassaemia trait carriers; in some communities and states this rises to 8–15%), making haemoglobin electrophoresis interpretation a high-prevalence, high-impact skill for every Indian paediatrician.
Indications for requesting haemoglobin electrophoresis:
- Microcytic anaemia not responding to 4–6 weeks of iron therapy (rule out thalassaemia trait or disease)
- Family history of thalassaemia, sickle cell disease, or haemoglobinopathy in parents or siblings
- Chronic haemolytic anaemia with target cells, NRBCs, or splenomegaly in the absence of IDA
- Newborn screening programme (where available) — mandatory in high-prevalence areas
- Pre-marital or antenatal carrier screening in families from high-prevalence regions (Maharashtra, Gujarat, Rajasthan for sickle cell; Punjab, Bengal for beta-thalassaemia)
- Elevated HbF detected incidentally on automated analysers
Interpreting the report — key fraction patterns:
| Pattern | HbA | HbA2 | HbF | HbS | Clinical interpretation |
|---|---|---|---|---|---|
| Normal adult | ~97% | 2–3.5% | <1% | 0 | Normal |
| Beta-thalassaemia trait | ~90–94% | >3.5% | slight ↑ | 0 | Carrier — genetic counselling |
| Beta-thalassaemia major | absent or trace | ↑ | ↑↑ (70–90%) | 0 | Transfusion-dependent; refer |
| Sickle cell disease (HbSS) | absent | ~2–3.5% | variable ↑ | ~80–95% | Vaso-occlusive disease |
| Sickle cell trait (HbAS) | ~50–60% | ~2–3.5% | <1% | ~35–45% | Carrier; usually asymptomatic |
| HbSC disease | absent | — | variable | ~50% | HbC ~50%; milder than HbSS |
| HPFH (Hereditary Persistence of HbF) | ↓ | normal | ↑↑ (15–30%) | 0 | Benign; mild or no anaemia |
Critical points for report interpretation:
- HbA2 >3.5% = beta-thalassaemia trait — this threshold is absolute. Iron deficiency can lower HbA2 (masking thalassaemia trait) — always correct iron deficiency before interpreting electrophoresis.
- No HbA band + HbS dominant = HbSS (not HbS trait). Sickle cell trait (HbAS) retains a prominent HbA band.
- Elevated HbF alone in an older child (beyond 6 months, when physiological HbF has declined) = beta-thalassaemia major or HPFH — distinguish by severity of anaemia and other fractions.
- Action after the report: beta-thalassaemia trait → parent testing → genetic counselling; thalassaemia major → register for regular transfusion programme, chelation; HbSS → penicillin prophylaxis, NIS immunisation, folate supplementation, hydroxyurea if indicated.
Haemoglobin Electrophoresis Band Patterns
SELF-CHECK
A 9-year-old girl has haemoglobin 9.2 g/dL, MCV 65 fL, normal RDW. Her haemoglobin electrophoresis shows: HbA 93%, HbA2 4.1%, HbF 0.8%, HbS absent. What is the most likely diagnosis and the single most important next step?
A. Iron deficiency anaemia — start oral iron supplementation
B. Beta-thalassaemia trait — arrange parental electrophoresis for genetic counselling
C. Alpha-thalassaemia trait — no further action needed
D. Megaloblastic anaemia — check serum B12 and folate levels
Reveal Answer
Answer: B. Beta-thalassaemia trait — arrange parental electrophoresis for genetic counselling
The combination of microcytic anaemia with normal RDW (uniform microcytosis, minimal anisocytosis) plus HbA2 >3.5% (here 4.1%) on electrophoresis is diagnostic of beta-thalassaemia trait. The MCV is low but the RDW is normal — this IDA-vs-thalassaemia distinction (IDA has high RDW, thalassaemia trait has normal RDW) is a key differentiating feature. The most important next step is parental electrophoresis to assess the risk of a thalassaemia-major child (if both parents are carriers, 25% risk per pregnancy). Iron supplementation alone would be incorrect and potentially harmful without addressing the underlying haemoglobinopathy.
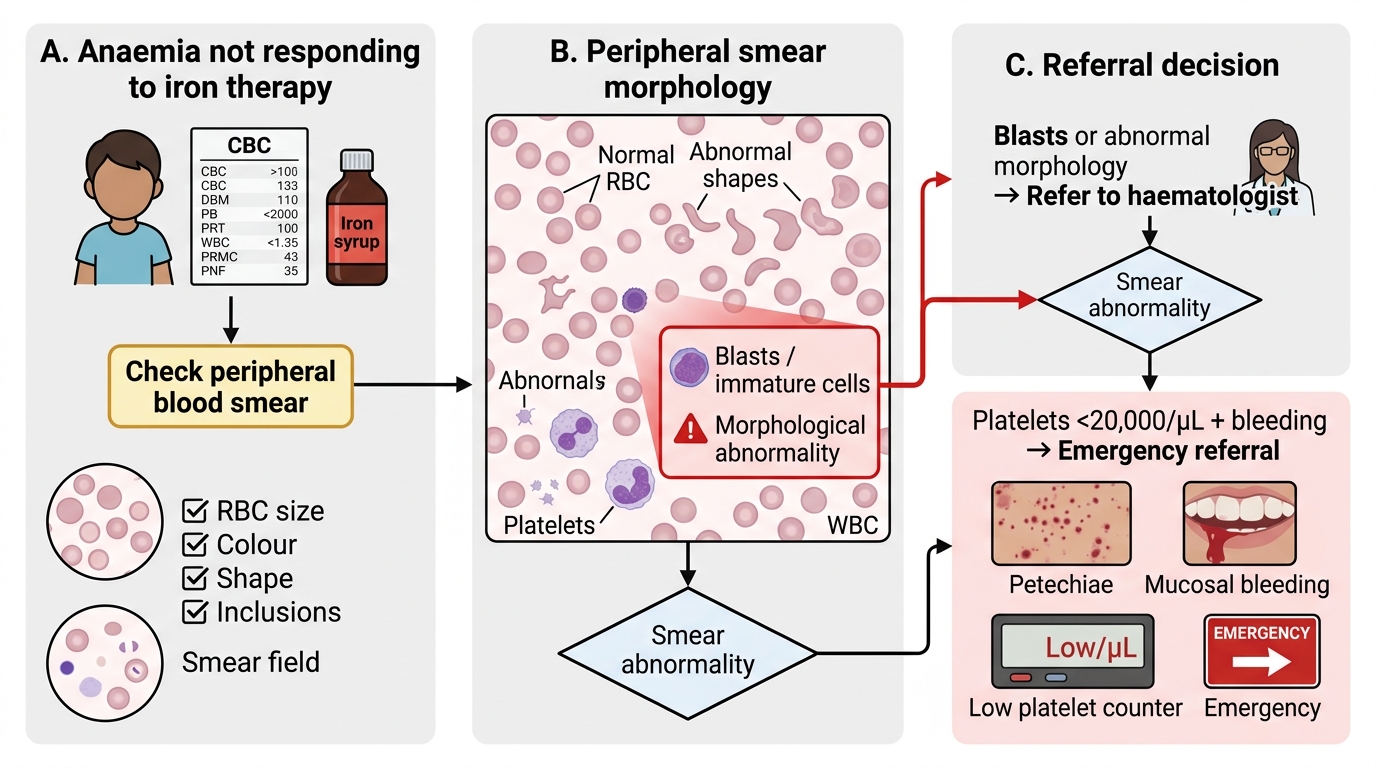
Applied Practice: Referral Criteria and Splenectomy Decisions
Referral criteria for haematological conditions define the threshold at which a child's haematological problem exceeds the management capacity of a primary or secondary care provider and requires the expertise of a paediatric haematologist at a tertiary centre. Over-referral wastes limited tertiary-centre resources and delays care for truly critical patients; under-referral is clinically dangerous and results in late diagnosis of curable or controllable conditions such as leukaemia, thalassaemia major, and aplastic anaemia. The art of referral decision-making rests on applying clear clinical and laboratory triggers rather than gut feeling — and on distinguishing the same-day emergency (blast cells, febrile neutropenia, acute sequestration) from the planned elective referral (iron-unresponsive anaemia, confirmed haemoglobinopathy for programme enrolment). In the Indian health system context, where tertiary paediatric haematology centres are concentrated in large cities, a timely referral decision made at a district-level primary care provider or community health centre can be the difference between a child who reaches a centre in time for curative chemotherapy and one who presents in end-stage disease. The IAP and AIIMS national guidelines provide the framework below; every paediatrician at the primary level must know these triggers and be able to communicate them clearly to families.
Applied practice also requires understanding splenectomy — a surgical procedure that has lifelong haematological and immunological implications for a child. The decision to splenectomise is never taken lightly: the spleen is the primary organ for filtration of encapsulated bacteria and for early IgM antibody responses in the unprimed child, and its removal creates a permanent susceptibility to overwhelming sepsis. The surgeon, the paediatric haematologist, and the paediatrician must jointly consider the indications, optimal timing by age, and mandatory pre- and post-operative precautions before proceeding. This section builds the competency to enumerate both the referral criteria and the splenectomy indications and precautions that the NMC CBME framework (PE26.15, PE26.17) requires.
Refer urgently (same-day transfer):
- Hb <6 g/dL in any age group, especially with cardiac compromise (tachycardia, respiratory distress, altered sensorium)
- Blast cells on peripheral smear — suspicion of acute leukaemia; do not wait for BMA at primary level
- Platelets <10,000/µL, or <20,000/µL with active bleeding (petechiae, mucosal bleeds, epistaxis not controllable)
- ANC <200/µL (profound neutropenia) with fever — febrile neutropenia is an oncological emergency
- Suspected DIC (prolonged PT + aPTT + low fibrinogen + clinical haemorrhage)
- Sickling crisis with stroke signs or acute chest syndrome (dyspnea + chest pain + hypoxia in HbSS)
Refer electively (within 1–2 weeks):
- Anaemia Hb 6–8 g/dL not responding to 4–6 weeks of appropriate iron therapy
- Confirmed thalassaemia major (no HbA on electrophoresis) — for enrolment in transfusion-chelation programme
- Confirmed HbSS — for hydroxyurea assessment, prophylaxis, and NIS catch-up
- Hereditary spherocytosis with symptomatic anaemia or frequent haemolytic crises — for splenectomy assessment
- Unexplained thrombocytopenia not responding to IVIG in ITP after 6 months
Indications for splenectomy in children:
Splenectomy is an irreversible intervention that eliminates the immune functions of the spleen; thus it is never undertaken lightly in the paediatric age group. The spleen is the primary site of filtration of encapsulated organisms (Streptococcus pneumoniae, Haemophilus influenzae type b, Neisseria meningitidis), and its removal creates overwhelming post-splenectomy infection (OPSI) risk — particularly in children under 5 years, in whom the risk of fatal OPSI can reach 50–80 times that of an age-matched intact patient.
Indications accepted in paediatric practice:
• Hereditary spherocytosis — the clearest paediatric indication; splenectomy is curative of the haemolysis (it does not fix the membrane defect but removes the site of RBC destruction). Deferred until age ≥6 years to allow immunological maturation.
• Thalassaemia major with hypersplenism — when transfusion requirements exceed 200–250 mL/kg/year of packed cells (indicating splenic sequestration and hypersplenism), splenectomy reduces transfusion frequency. Deferred until age ≥5–6 years.
• ITP (immune thrombocytopenic purpura) — chronic ITP (>12 months) refractory to corticosteroids and IVIG; second-line alternative to rituximab and TPO-agonists in children ≥5 years.
• Sickle cell disease — symptomatic hypersplenism, acute sequestration crises (repeated), massive splenomegaly causing compression symptoms.
• Splenic sequestration crisis — life-threatening acute crisis in sickle cell disease with massive rapid splenic enlargement and Hb drop >2 g/dL from baseline.
Precautions after splenectomy — mandatory:
1. Vaccination BEFORE surgery (ideally ≥2 weeks before elective splenectomy, or as soon as stable after emergency splenectomy): Pneumococcal conjugate vaccine (PCV13 + PPSV23), Hib conjugate vaccine, meningococcal conjugate vaccine (MenACWY + MenB). These align with the National Immunization Schedule and the WHO position paper on asplenia.
2. Post-splenectomy penicillin prophylaxis: oral phenoxymethylpenicillin (penicillin V) 125 mg twice daily for children <5 years; 250 mg twice daily for ≥5 years — continued for at least 2 years post-splenectomy and until age 5 in all children; some centres continue lifelong.
3. Education of family: child and caregivers must know that any fever ≥38°C is a medical emergency and must be seen within 2 hours (OPSI can progress to septic shock within 12–24 hours). Provide a 'splenectomy alert' card.
4. Medical alert identification (bracelet or card).
5. Annual influenza vaccination.
Paediatric Haematology Referral Decision Flowchart
| Condition | Splenectomy indication | Minimum age | Key precautions |
|---|---|---|---|
| Hereditary spherocytosis | Symptomatic haemolysis, gallstones | ≥6 yr | PCV13 + PPSV23 + Hib + MenACWY; penicillin prophylaxis |
| Thalassaemia major | Hypersplenism (transfusions >200–250 mL/kg/yr) | ≥5–6 yr | Same vaccinations; chelation unchanged |
| Chronic ITP | Refractory >12 months, after IVIG + steroids fail | ≥5 yr | Vaccinations before surgery |
| Sickle cell disease | Splenic sequestration crises (recurrent), hypersplenism | Individual | Pneumococcal + Hib + MenACWY; penicillin prophylaxis |