Page 4 of 33
PE12.3 | Vitamin D — SDL Guide
Learning Objectives
- Describe the causes of Vitamin D deficiency in children including nutritional, environmental and metabolic factors
- Identify the clinical features of nutritional rickets including skeletal deformities, systemic signs and complications
- Classify rickets and distinguish nutritional from metabolic subtypes
- Interpret the biochemical and radiological findings in active rickets
- Prescribe the correct treatment for nutritional rickets (stoss therapy and daily therapy) and state the prophylaxis dose
- Recognise the clinical features and management of Vitamin D toxicity (hypervitaminosis D)
INSTRUCTIONS
Nutritional rickets remains a common paediatric problem in India, with both Vitamin D deficiency and dietary calcium deficiency contributing to the burden. The disease causes painful, disfiguring skeletal deformities and, in infants, dangerous hypocalcaemic seizures. Treatment is highly effective when initiated early, and the disease is entirely preventable with simple supplementation from birth. This module covers the disease arc — clinical presentation, pathophysiology, diagnosis, and management — equipping you to recognise, treat, and prevent rickets in clinical practice.
References
- Ghai Essential Pediatrics, 9th ed., Ch 9 (Vitamins and Minerals — Vitamin D) (textbook)
- Nelson Textbook of Pediatrics, 21st ed., Ch 63 (Vitamin D Deficiency and Rickets) (textbook)
- IAP Consensus Statement on Vitamin D Deficiency in Childhood and Adolescence (2017) (guideline)
- WHO/UNICEF Technical Note on Nutritional Rickets: Summary of Deliberations (2016) (guideline)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A 14-month-old boy is brought to the outpatient department with his mother, who is worried that he is 'not walking properly.' He was exclusively breastfed until 10 months, is vegetarian, and rarely goes outdoors. On examination he is irritable, his anterior fontanelle is still open, and you notice prominent widening at the wrists. When you observe his gait, you see bilateral bowing of the legs. Blood tests sent from the primary health centre show serum calcium 7.2 mg/dL, phosphate 2.8 mg/dL, and alkaline phosphatase 980 U/L. What is the diagnosis? What single curative intervention is available, and how would you prevent this condition in his younger sibling, born last month?
WHY THIS MATTERS
Rickets is the softening and weakening of bones in children caused by inadequate mineralisation during active bone growth — principally due to Vitamin D deficiency or dietary calcium deficiency. It is among the most common nutritional deficiency diseases seen in Indian paediatric practice, with a particular predilection for breastfed infants in low-sunlight environments, dark-skinned children, and those with poor dietary diversity. The consequences extend beyond orthopaedic deformity: in infants, severe hypocalcaemia from rickets causes life-threatening seizures and laryngospasm. Untreated skeletal deformity (genu varum, coxa vara, pelvic distortion) can persist into adult life, causing chronic disability, difficult labour in females, and chronic pain. Critically, the disease is both eminently preventable (400 IU of Vitamin D daily from birth) and highly treatable once diagnosed. Understanding the calcium-phosphate-PTH-Vitamin D axis and the radiological hallmarks of rickets is essential for every doctor working in India.
RECALL
Activate your prior knowledge of calcium homeostasis from Physiology and Biochemistry. Recall that serum calcium is regulated by three hormones: Parathyroid Hormone (PTH), Vitamin D (calcitriol), and calcitonin. PTH is released in response to hypocalcaemia and acts on bone (calcium resorption), kidney (calcium reabsorption, phosphate excretion, and stimulation of 1-alpha-hydroxylase), and intestine (indirectly via calcitriol). The active form of Vitamin D — 1,25-dihydroxyvitamin D (calcitriol) — is synthesised in the kidney by 1-alpha-hydroxylase, which converts the hepatic intermediate 25-hydroxyvitamin D (calcidiol). These steps are the foundation for understanding why Vitamin D deficiency impairs bone mineralisation: without calcitriol, intestinal calcium absorption falls, PTH rises, bone matrix is incompletely mineralised (osteoid accumulates), and the growth plate expands abnormally.
Clinical Presentation of Rickets
Nutritional rickets typically presents in children between 6 months and 3 years of age — the period of most rapid skeletal growth when the demand for mineralisation is highest. The disease presents with a characteristic combination of skeletal deformities, systemic features of hypocalcaemia, and signs of delayed development. The clinical picture varies depending on the child's age and which part of the skeleton is growing most rapidly at the time: skull changes predominate in early infancy, chest deformities in the first year, and lower-limb bowing becomes prominent once the child begins to bear weight. This age-related variation means that the same diagnosis may present very differently in a 3-month-old infant (craniotabes, hypocalcaemic seizures) compared to an 18-month-old toddler (genu varum, wrist widening). A systematic head-to-toe examination is essential to capture the full spectrum.
Skeletal manifestations are the most recognisable features and vary with the region of skeleton most actively growing at the time of deficiency:
Skull and head:
• Craniotabes: softening of the occipital and parietal bones, felt as a 'ping-pong ball' on pressing; earliest sign in infants <6 months (also seen in premature infants without rickets — not pathognomonic alone)
• Frontal bossing: prominence of the frontal and parietal bones due to excessive osteoid
• Delayed fontanelle closure: anterior fontanelle remains open beyond 18 months
• Delayed dentition
Chest:
• Rachitic rosary: beading at the costochondral junctions (soft, palpable, non-tender beads)
• Harrison's sulcus: horizontal depression at the line of diaphragmatic attachment — due to softened rib cage being pulled inward by the diaphragm
• Pigeon chest (pectus carinatum) or bell-shaped chest
Limbs:
• Widening of wrists and ankles: metaphyseal expansion — a very useful clinical sign, easy to elicit
• Genu varum (bow legs) in walking children (lower limb weight-bearing forces curve the soft bones)
• Genu valgum (knock knees) — less common, seen more in older children
• Coxa vara — bowing at the hip
Spine:
• Kyphoscoliosis in older children
Systemic features reflect hypocalcaemia:
• Hypocalcaemic seizures — the most dangerous complication; can be the presenting feature in infants; tetany (Chvostek's and Trousseau's signs positive)
• Laryngospasm — stridor from hypocalcaemic laryngeal spasm; may mimic croup
• Muscle hypotonia — causes delayed motor milestones (sitting, standing, walking)
• Irritability and poor feeding
• Recurrent infections — Vitamin D has immunomodulatory functions
• Growth retardation — both height and weight affected in chronic disease
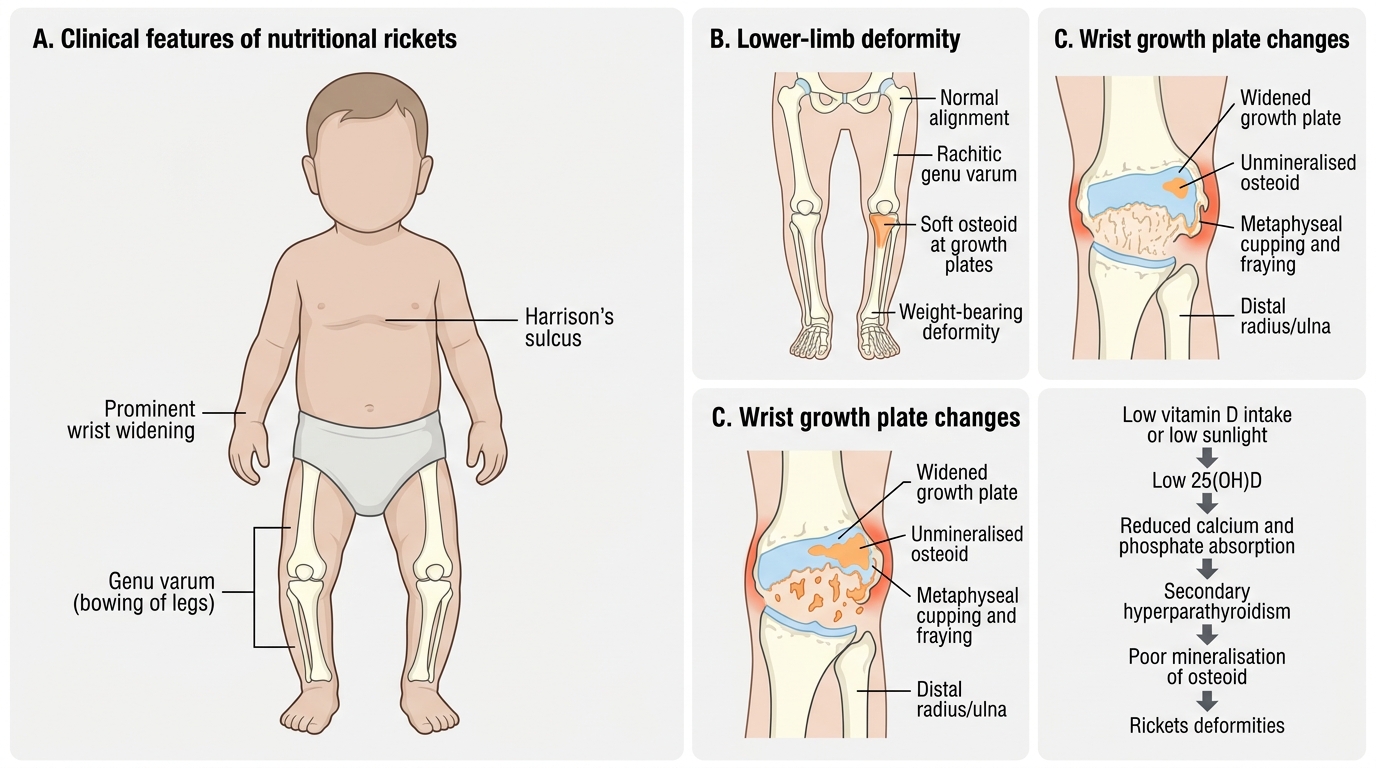
Clinical Features and Pathophysiology of Nutritional Rickets
Pathophysiology and Aetiology of Vitamin D Deficiency
Understanding the metabolism of Vitamin D illuminates both its multiple causes and the rationale for treatment. Vitamin D exists in two dietary forms: Vitamin D3 (cholecalciferol), synthesised in the skin from 7-dehydrocholesterol under the influence of UV-B radiation (wavelength 290–315 nm), and Vitamin D2 (ergocalciferol), derived from fungal and plant sources. Vitamin D is not biologically active in either of these forms — it must be converted in two sequential steps, first in the liver and then in the kidney, before it can regulate calcium and phosphate homeostasis. This two-step activation means that disease at either step (liver disease, renal disease) can produce clinical Vitamin D deficiency even when dietary intake and sun exposure are adequate, and explains why renal failure causes secondary hyperparathyroidism and metabolic bone disease. In nutritional rickets, the problem lies upstream — inadequate substrate entering the metabolic pathway — rather than in the conversion enzymes themselves. Both forms undergo two sequential hydroxylation steps before becoming biologically active:
- Hepatic 25-hydroxylation: Vitamin D3/D2 is hydroxylated by hepatic CYP2R1 (25-hydroxylase) to form 25-hydroxyvitamin D [25(OH)D, calcidiol] — the principal circulating storage form, measured to assess Vitamin D status.
- Renal 1-alpha-hydroxylation: 25(OH)D is converted in the proximal tubular cells of the kidney by CYP27B1 (1-alpha-hydroxylase) to 1,25-dihydroxyvitamin D [1,25(OH)2D, calcitriol] — the active hormone. This step is stimulated by PTH, hypophosphataemia, and hypocalcaemia; it is inhibited by fibroblast growth factor-23 (FGF-23) and by renal disease.
Calcitriol's actions:
• Increases intestinal absorption of calcium and phosphate (principal action)
• Stimulates renal calcium reabsorption
• Acts on osteoblasts to promote bone mineralisation
• Has immunomodulatory effects (reduces susceptibility to infections, autoimmune diseases)
Causes of nutritional Vitamin D deficiency rickets in India:
• Inadequate UV-B exposure: exclusive indoor living, air pollution reducing UV-B penetration, northern latitudes in winter, covering of the body (cultural dress practices, purdah)
• Exclusive breastfeeding without supplementation: breast milk contains very little Vitamin D (15–50 IU/L regardless of maternal status); an exclusively breastfed infant receives ~1–3 IU/kg/day — far below the 400 IU/day requirement
• Dark skin pigmentation: melanin reduces UV-B penetration; children with darker skin need more sun exposure to synthesise equivalent Vitamin D
• Prematurity: inadequate placental transfer and limited postnatal sun exposure
• Malabsorption syndromes: coeliac disease, cholestatic liver disease, short bowel syndrome impair absorption of fat-soluble vitamins
• Calcium deficiency (calcium-deficiency rickets): seen in parts of South India and sub-Saharan Africa where dietary calcium intake is extremely low despite adequate Vitamin D; the mechanism is secondary hyperparathyroidism driven by low calcium, resulting in phosphaturia and undermineralisation
Deficiency status (serum 25(OH)D):
• Deficiency: <20 ng/mL (<50 nmol/L)
• Insufficiency: 20–30 ng/mL
• Sufficient: >30 ng/mL
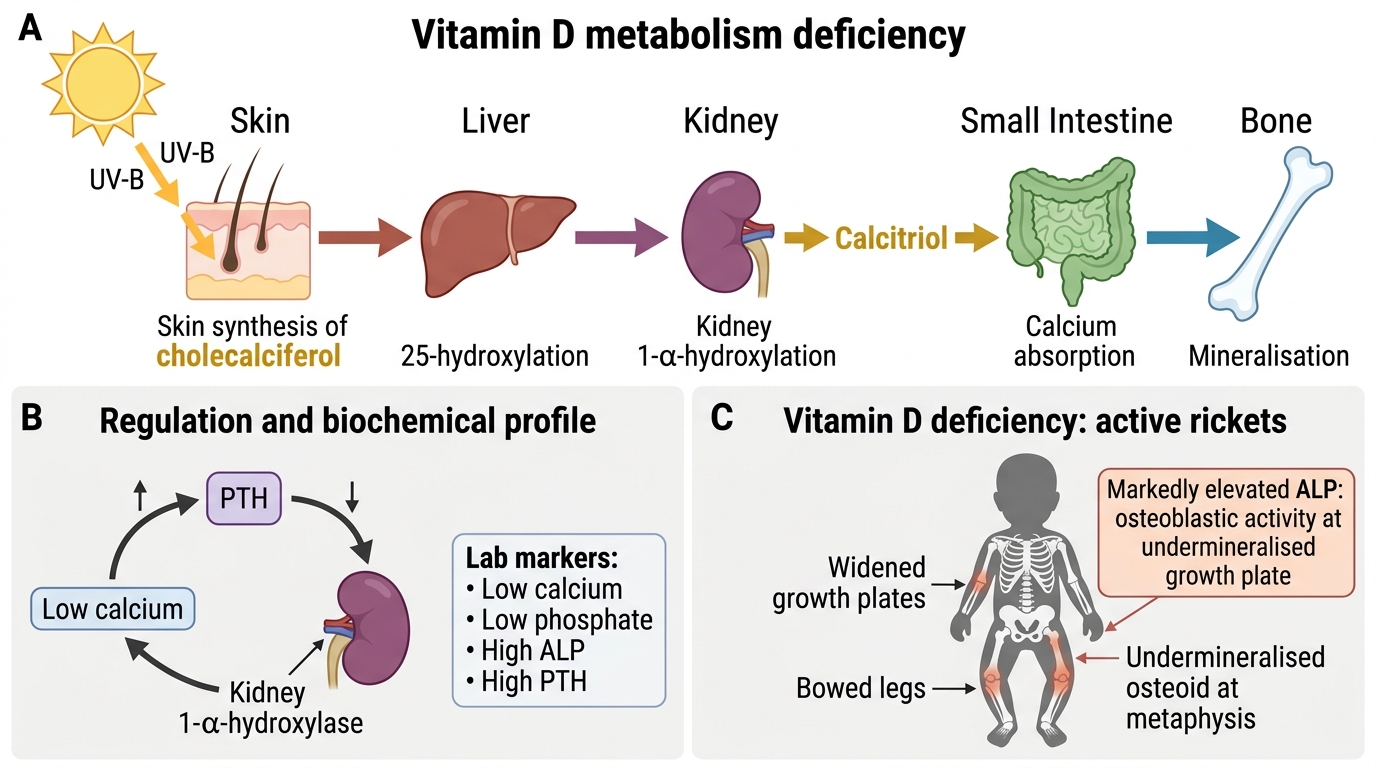
Vitamin D Metabolism and Deficiency Consequences
SELF-CHECK
An 18-month-old child presents with hypocalcaemic seizures. Investigations reveal: serum calcium 6.8 mg/dL (low), phosphate 2.5 mg/dL (low), ALP 1200 U/L (elevated), and PTH 180 pg/mL (elevated). Which biochemical finding most specifically confirms active rickets rather than simple dietary calcium deficiency alone?
A. Elevated alkaline phosphatase (ALP) reflecting increased osteoblastic activity at the undermineralised growth plate
B. Low serum calcium indicating insufficient dietary calcium intake
C. Elevated PTH indicating secondary hyperparathyroidism from any calcium-depleting cause
D. Low serum phosphate indicating renal phosphate wasting from any cause
Reveal Answer
Answer: A. Elevated alkaline phosphatase (ALP) reflecting increased osteoblastic activity at the undermineralised growth plate
Markedly elevated ALP is the most specific biochemical marker of active rickets — it reflects intense osteoblastic activity at the undermineralised growth plate where osteoid is accumulating. All four abnormalities (low Ca, low PO4, high ALP, high PTH) together constitute the 'rickets biochemical profile', but ALP is most specifically tied to the bone disease process. Low calcium and elevated PTH can occur in simple dietary calcium deficiency or other causes of hypocalcaemia; low phosphate can result from renal wasting. ALP elevation in the context of skeletal disease is the hallmark.
Diagnosis and Investigation of Rickets
The diagnosis of rickets is clinical and radiological, supported by characteristic biochemical findings. In the community setting, a confident clinical diagnosis can often be made on history and physical examination alone, with investigations confirming the severity and guiding treatment. A combination of skeletal signs (rachitic rosary, wrist widening, genu varum) in a child with known risk factors (exclusively breastfed without supplementation, limited sunlight exposure) is highly suggestive. However, investigations are essential for three reasons: to confirm the diagnosis when clinical signs are equivocal, to distinguish nutritional Vitamin D deficiency from metabolic subtypes (renal osteodystrophy, Vitamin D-dependent rickets, hereditary hypophosphataemic rickets), and to establish a baseline for monitoring treatment response. The biochemical profile of nutritional rickets has a characteristic pattern that reflects the downstream consequences of calcitriol deficiency on calcium-phosphate-PTH homeostasis. Understanding this pattern allows the clinician to both confirm the diagnosis and infer the underlying mechanism.
Biochemical profile in active nutritional rickets:
| Investigation | Expected finding in active nutritional rickets |
|---|---|
| Serum calcium | Low or low-normal (7–8.5 mg/dL) |
| Serum phosphate | Low (renal phosphate wasting driven by PTH) |
| Serum ALP | Markedly elevated (hallmark — reflects osteoblastic activity) |
| Serum PTH | Elevated (secondary hyperparathyroidism) |
| Serum 25(OH)D | Low (<20 ng/mL in nutritional Vitamin D deficiency) |
| Serum 1,25(OH)2D | May be low or raised (PTH stimulates 1-alpha-hydroxylase) |
| Urinary calcium | Very low (hypocalciuria — calcium is being retained) |
Radiological features — X-ray of wrist and knee (most informative):
The changes reflect failure of normal metaphyseal mineralisation:
• Cupping, fraying, and splaying of the metaphysis — the most characteristic sign; the normally sharp, straight metaphyseal edge becomes flared, irregular, and concave ('champagne glass' deformity)
• Widened growth plate — the zone of provisional calcification fails to mineralise
• Reduced bone density (osteopenia) — generalised thinning of cortices
• Delayed appearance of epiphyseal ossification centres
• In healing rickets: Looser's zones (pseudofractures) — translucent bands perpendicular to the bone cortex (more common in osteomalacia/adults)
• Codfish vertebrae (biconcave vertebral bodies) in severe spinal involvement
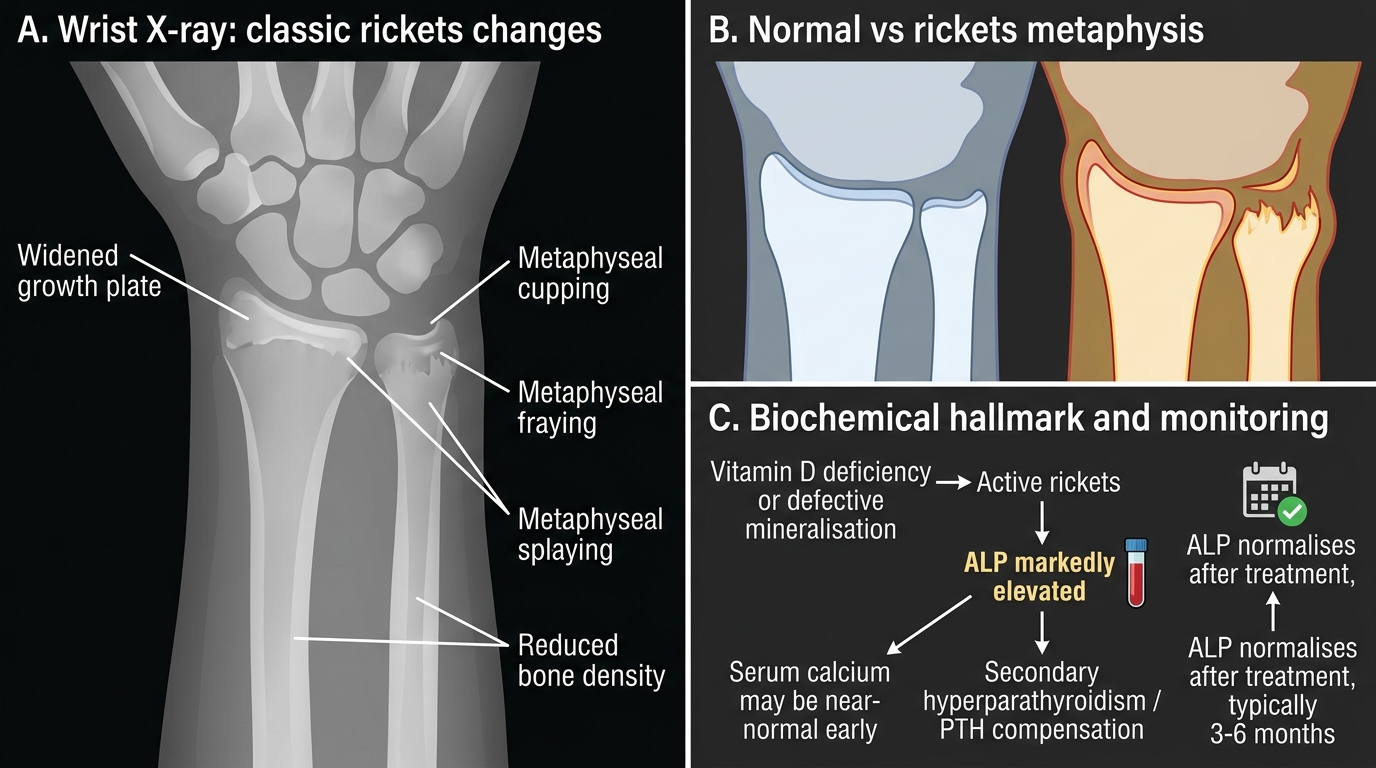
Wrist X-ray Changes and ALP Hallmark in Active Rickets
CLINICAL PEARL
The biochemical hallmark of active rickets is a markedly elevated serum alkaline phosphatase (ALP) — not hypocalcaemia. Serum calcium may actually be near-normal in early rickets because PTH-driven skeletal resorption partially compensates. ALP is elevated in virtually every case of active rickets and can be used to monitor treatment response: normalisation of ALP (typically 3–6 months after treatment) confirms biochemical healing. For monitoring, ALP is cheaper and more practical than repeated 25(OH)D levels.