Page 1 of 21
PA16.1 | Haemolytic Anaemia — Definition, Classification & Lab Markers — SDL Guide
Learning Objectives
- Define haemolysis and haemolytic anaemia, and distinguish compensated from decompensated states.
- Classify haemolytic anaemias using three parallel frameworks: site (intravascular vs extravascular), cause (hereditary vs acquired), and defect location (intracorpuscular vs extracorpuscular).
- Describe the general clinical features of haemolytic anaemia and explain their pathophysiological basis.
- Identify the key haematologic indices and laboratory markers of haemolysis and interpret their significance.
- Recognise the categories of hereditary and acquired haemolytic anaemias as a foundation for SDL2 and SDL3.
INSTRUCTIONS
Red cell destruction is one of the commonest causes of anaemia you will encounter in clinical medicine. This module builds the conceptual scaffolding — definitions, classification frameworks, clinical features and laboratory hallmarks — that underpins every specific haemolytic disorder you will study. A firm grasp of the three classification axes and the laboratory marker map will allow you to reason systematically about any patient presenting with haemolysis, from sickle-cell disease to autoimmune haemolytic anaemia.
References
- Robbins & Cotran Pathologic Basis of Disease, 10th ed., Ch 14 (Red Cell and Bleeding Disorders) (textbook)
- Harsh Mohan's Textbook of Pathology, 8th ed., Ch 12 (Disorders of Red Blood Cells) (textbook)
- Harrison's Principles of Internal Medicine, 21st ed., Ch 100 (Haemolytic Anaemias) (textbook)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A 19-year-old boy from Tamil Nadu presents with yellow eyes since childhood, recurrent dark urine during fevers, and a palpable spleen that his mother says has been there 'since he was a baby.' His haemoglobin is 8 g/dL, reticulocyte count is 14%, and his peripheral smear shows small spherical red cells with no central pallor. His father had his gallbladder removed at age 32 for pigment stones.
This clinical picture — jaundice, splenomegaly, dark urine, a reticulocyte surge, and a positive family history — is haemolysis written in plain language. By the end of this SDL, you will be able to decode every one of these findings from first principles.
WHY THIS MATTERS
Haemolytic anaemias account for roughly 5–10% of all anaemias presenting to medicine wards in India. They span genetics (sickle-cell disease, thalassaemia, G6PD deficiency — all disproportionately prevalent on the Indian subcontinent), autoimmunity (AIHA), and systemic disease (microangiopathic haemolytic anaemia in DIC, TTP, HUS). The classification framework you build here is your diagnostic map for all of these conditions. PA16.1 is the gateway competency for the entire haemolytic anaemia block (SDL1→SDL2→SDL3) and is directly examined in University theory and clinical case stations.
RECALL
Before proceeding, confirm you can answer these from Year-1:
• What is the normal lifespan of a red blood cell, and where are aged red cells destroyed?
• What are the two main steps in haem catabolism, and at which step is unconjugated (indirect) bilirubin produced?
• What is the role of haptoglobin in normal red cell turnover?
• Name three structural components of the red cell membrane relevant to cell deformability.
If any of these feel uncertain, revisit your Year-1 Physiology (RBC physiology) and Biochemistry (haem catabolism) notes before continuing — this SDL builds directly on those foundations.
Defining Haemolysis and Haemolytic Anaemia

Hemolysis Spectrum: From Normal RBC Lifespan to Hemolytic Anemia
Haemolysis is the premature destruction of red blood cells before the end of their normal 120-day lifespan. It may be physiological (senescent cells destroyed daily in the spleen, liver and bone marrow) or pathological (accelerated destruction due to intrinsic or extrinsic red cell defects).
Haemolytic anaemia develops when the rate of red cell destruction exceeds the compensatory capacity of the bone marrow. The bone marrow normally has a six- to eight-fold reserve — it can increase erythropoiesis six- to eight-fold in response to a haemolytic stimulus. When destruction is moderate, the marrow may fully compensate, maintaining a normal or near-normal haemoglobin. When destruction outpaces even maximal marrow output, clinical anaemia supervenes.
Two key states arise from this relationship:
• Compensated haemolytic state: RBC lifespan is shortened (e.g., 30–60 days instead of 120) but the accelerated marrow output keeps haemoglobin within or near the normal range. The patient may be mildly jaundiced and splenomegalic but not anaemic. This state can persist for years — hereditary spherocytosis is a classic example.
• Decompensated haemolytic state (haemolytic anaemia): Destruction rate exceeds maximal marrow compensation. Haemoglobin falls. Clinical anaemia — fatigue, pallor, dyspnoea on exertion — becomes manifest. Intercurrent infections, folate deficiency, or an aplastic crisis (parvovirus B19) can tip a compensated state into decompensation.
Classification Axis 1 — Site of Haemolysis: Intravascular vs Extravascular
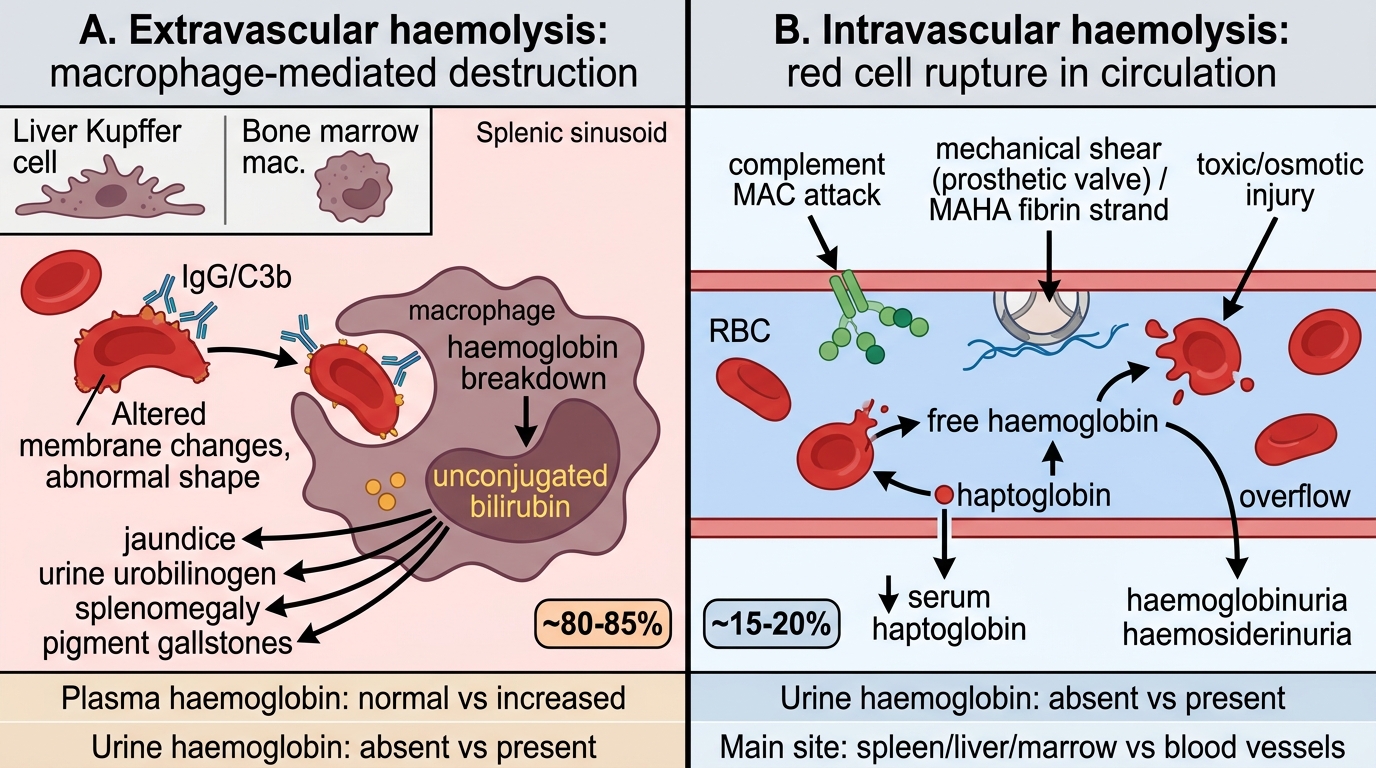
Intravascular vs Extravascular Haemolysis
The most mechanistically important classification axis divides haemolysis by where the red cell is destroyed.
Extravascular haemolysis (the majority, ~80–85% of all haemolysis)
• Site: Mononuclear phagocyte system — primarily splenic sinusoids, but also Kupffer cells in the liver and bone marrow macrophages.
• Mechanism: Red cells with altered membrane proteins, abnormal shapes, or surface-bound IgG/C3b are recognised and engulfed by macrophages. The cell is phagocytosed intact; haemoglobin is catabolised inside the macrophage.
• Lab signature: ↑unconjugated (indirect) bilirubin → jaundice; ↑urine urobilinogen; normal plasma haemoglobin; no haemoglobinuria; splenomegaly (from macrophage hyperplasia and work hypertrophy); pigment gallstones (calcium bilirubinate) from chronic bilirubin overload.
• Examples: Hereditary spherocytosis, warm AIHA, sickle-cell disease (most haemolysis extravascular).
Intravascular haemolysis (minority, ~15–20%)
• Site: Inside blood vessels — red cell membrane ruptures directly within the circulation.
• Mechanism: Complement-mediated lysis (MAC, e.g., PNH), mechanical destruction (prosthetic heart valves, MAHA), osmotic or toxic lysis, or severe immune haemolysis.
• Lab signature: ↑plasma free haemoglobin → binds haptoglobin → ↓serum haptoglobin; when haptoglobin is saturated, free Hb passes glomerulus → haemoglobinuria (red/brown urine); chronic intravascular haemolysis → iron deposition in renal tubular cells → urinary haemosiderin (Prussian blue-positive tubular cells in urine sediment); ↑LDH (released from lysed cells); methaemalbuminaemia.
• Examples: PNH, G6PD deficiency during oxidant crisis, march haemoglobinuria, MAHA (DIC, TTP, HUS), ABO-incompatible transfusion reaction.
| Feature | Extravascular | Intravascular |
|---|---|---|
| Site | Spleen/liver/marrow macrophages | Blood vessel lumen |
| Free plasma Hb | Normal/slight ↑ | Markedly ↑ |
| Haptoglobin | Mildly ↓ | Markedly ↓ |
| Haemoglobinuria | Absent | Present (severe) |
| Urinary haemosiderin | Absent | Present (chronic) |
| Jaundice | Prominent | Mild–moderate |
| Splenomegaly | Common | Variable |
IMPORTANT: Both types share ↑reticulocyte count, ↑LDH, and ↑unconjugated bilirubin — but the magnitude and accompanying features distinguish them.

Intravascular vs Extravascular Hemolysis Pathways
CLINICAL PEARL
The 'port-wine urine' clue: A patient with red-brown urine after a cold exposure (PNH, cold agglutinin disease) or strenuous exertion (march haemoglobinuria, G6PD crisis) is almost certainly experiencing intravascular haemolysis until proven otherwise. Check a urine dipstick — haemoglobin is detected as 'blood' by the peroxidase reaction. If microscopy shows no red cells but the dipstick is positive, haemoglobinuria (or myoglobinuria — check serum CK) is the answer, not haematuria. This one clinical observation directs you immediately to the intravascular haemolysis pathway.
Classification Axis 2 — Cause: Hereditary vs Acquired

Hemolytic Anemias: Hereditary vs Acquired Causes
The second axis asks why the red cell is being destroyed prematurely.
Hereditary (congenital) haemolytic anaemias: Due to inherited defects in the red cell itself. These are almost always intracorpuscular (see Axis 3). They typically present in childhood or early adulthood, are lifelong, and often show a positive family history. They include:
• Membrane defects: Hereditary spherocytosis, hereditary elliptocytosis
• Enzyme defects: G6PD deficiency, pyruvate kinase deficiency
• Haemoglobin defects: Sickle-cell disease, the thalassaemias
Acquired haemolytic anaemias: Develop in an individual with previously normal red cells due to an extrinsic pathological process. They may be intracorpuscular (e.g., PNH — a somatic mutation in a haemopoietic stem cell) or extracorpuscular (the majority). Acquired causes include immune (warm AIHA, cold AIHA), microangiopathic (DIC, TTP, HUS, pre-eclampsia), infection (malaria, Clostridium), drugs and chemicals, hypersplenism, and transfusion reactions.
Clinical clue: A previously well adult who develops haemolytic anaemia almost certainly has an acquired cause — systemic disease, autoimmunity, or a drug. A child or young adult with a lifelong history and a positive family history almost certainly has an inherited cause.